Phenoconversion of Cytochrome P450 Metabolism: A Systematic Review



1
Department of Clinical Pharmacy & Toxicology, Leiden University Medical Center, 2333 ZA Leiden, The Netherlands
2
Leiden Network for Personalised Therapeutics, Leiden University Medical Center, 2333 ZA Leiden, The Netherlands
3
Division of BioTherapeutics, Leiden Academic Centre for Drug Research (LACDR), Leiden University, 2333 CC Leiden, The Netherlands
*
Author to whom correspondence should be addressed.
J. Clin. Med. 2020, 9(9), 2890; https://doi.org/10.3390/jcm9092890
Submission received: 3 August 2020
/
Revised: 3 September 2020
/
Accepted: 4 September 2020
/
Published: 7 September 2020
(This article belongs to the Special Issue Advances and Challenges in Pharmacogenomics)
Abstract
:Phenoconversion is the mismatch between the individual’s genotype-based prediction of drug metabolism and the true capacity to metabolize drugs due to nongenetic factors. While the concept of phenoconversion has been described in narrative reviews, no systematic review is available. A systematic review was conducted to investigate factors contributing to phenoconversion and the impact on cytochrome P450 metabolism. Twenty-seven studies met the inclusion criteria and were incorporated in this review, of which 14 demonstrate phenoconversion for a specific genotype group. Phenoconversion into a lower metabolizer phenotype was reported for concomitant use of CYP450-inhibiting drugs, increasing age, cancer, and inflammation. Phenoconversion into a higher metabolizer phenotype was reported for concomitant use of CYP450 inducers and smoking. Moreover, alcohol, pregnancy, and vitamin D exposure are factors where study data suggested phenoconversion. The studies reported genotype–phenotype discrepancies, but the impact of phenoconversion on the effectiveness and toxicity in the clinical setting remains unclear. In conclusion, phenoconversion is caused by both extrinsic factors and patient- and disease-related factors. The mechanism(s) behind and the extent to which CYP450 metabolism is affected remain unexplored. If studied more comprehensively, accounting for phenoconversion may help to improve our ability to predict the individual CYP450 metabolism and personalize drug treatment.
1. Introduction
In the last decade, pharmacogenetics-informed drug dosing has progressively moved into clinical practice for various drug–gene combinations [1,2]. For more than 100 drug–gene pairs, guidelines to manage drug treatment based on genetic test results are available from the Dutch Pharmacogenetics Working Group (DPWG) and the Clinical Pharmacogenetics Implementation Consortium (CPIC) [3,4]. However, while there is an ever-increasing body of evidence supporting the clinical use of pharmacogenetics, the increased usage has also spawned awareness of its limitations.
DPWG and CPIC provide guidelines for genes encoding cytochrome P450 (CYP450) enzymes involved in human drug metabolism, including CYP1A2, CYP2D6, CYP2C9, CYP2C19, CYP3A4, and CYP3A5, which account for the metabolism of 70–80% of approved drugs [5]. Historically, based on observed variability in pharmacokinetics, genetic variants are categorized into five different predicted metabolizer phenotypes: normal metabolizers (NM), ultra-rapid metabolizers (UM), rapid metabolizers (RM), intermediate metabolizers (IM), and poor metabolizers (PM) [3,4].
In addition to genetics, many other factors influence drug metabolism, such as sex, age, nutritional condition, hormonal and diurnal influences, concomitant drug use, or (underlying) diseases [5]. Phenoconversion (Box 1 and Box 2) is the mismatch between the genotype-based prediction of CYP450-mediated drug metabolism and the true capacity of an individual to metabolize drugs (phenotype) due to nongenetic factors. Phenoconversion can result from the use of concomitant medication [6,7], comorbidities [7], or other factors such as smoking [8,9]. The effect of phenoconversion may differ per genotype. For example, concomitant use of an enzyme inhibitor in a genotypic CYP2D6 IM could result in a phenotypic CYP2D6 PM. By contrast, a CYP2D6 PM, lacking functional CYP2D6 enzyme, may be unaffected by the concomitant use of the same enzyme inhibitor since there is no enzyme to be inhibited and, hence, remain a CYP2D6 PM. However, in current clinical practice, genetic factors are ignored when interpreting drug–drug interactions (DDIs). Obviously, a genotype–phenotype mismatch could have significant consequences in clinical settings and may result in suboptimal treatment of patients. Moreover, phenoconversion might explain some of the conflicting results from pharmacogenetic studies and difficulties with replication of reported results [8,10].
While the concept of phenoconversion has been previously described in narrative reviews [9,11,12,13,14], no systematic review providing an overview of its impact on CYP450 metabolism is available. Therefore, we performed a systematic analysis of the factors contributing to phenoconversion and their impact in the context of CYP450 metabolism.
Box 1. Example of phenoconversion due to concomitant medication.
John is being treated with the CYP2D6 substrate nortriptyline (75 mg per day) for his depression. John was genotyped as CYP2D6*1/*5 and predicted to be a CYP2D6 intermediate metabolizer (IM) according to recent consensus recommendations [15]. Consequently, his maintenance dose was adjusted to 60% of the standard dose of nortriptyline, according to DPWG guidelines [3,4]. John has recently been diagnosed with arrhythmia and has, therefore, started therapy with the weak CYP2D6 inhibitor amiodarone [16]. Subsequently, John experienced nortriptyline-related side effects, such as tremor, hyperthermia, and tachycardia. John’s nortriptyline plasma concentrations resembled those found in a CYP2D6 PM phenotype. If John would have been genotyped as CYP2D6*1/*1, which is interpreted as a CYP2D6 NM phenotype, the drug interaction with amiodarone would have had a minimal or no effect on John’s CYP2D6 phenotype, and he would have remained a CYP2D6 NM or shifted to a CYP2D6 IM. This case shows that the effect of drug–drug interactions (DDI) on cytochrome p450 activity is highly dependent on the patient’s genotype.
Box 2. Example of phenoconversion due to comorbidity.
Suzan was treated with the CYP3A5 substrate tacrolimus for immunosuppression after kidney transplantation. She was genotyped as CYP3A5*1/*3, which is interpreted as a CYP3A5 IM phenotype [17]. Suzan has been successfully treated with 5 mg tacrolimus b.i.d. since her kidney transplant. However, she recently acquired an infection that caused the release of proinflammatory cytokines and increased levels of c-reactive protein (CPR) to 200 mg/L. Suzan began to experience tacrolimus-related size effects such as tremor, vomiting, and increased serum creatinine concentrations. Suzan’s tacrolimus plasma concentrations were found to be above the therapeutic range, which indicates the occurrence of phenoconversion from a CYP3A5 IM to PM.
2. Methods
2.1. Identification of Eligible Studies
For this systematic review, identification and selection of studies were performed according to the PICO method [18]. Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines were used to prepare the report [19]. PubMed was used to identify and extract all relevant papers until June 2020. Search terms consisted of phenoconversion and synonyms or explanations of this term, together with terms describing comedication, comorbidity, and other factors that were found to be related to phenoconversion, combined with terms for the different names of CYP450 enzymes. The full search string is provided in Appendix A. Reference lists from reviews were manually checked to identify relevant cross-references. Identified records were screened on title and abstract. Comments, editorials, narrative reviews, letters (without original data), abstracts, and publications in languages other than English were excluded.
The remaining records were assessed on their appropriateness for demonstrating phenoconversion, i.e., the mismatch between the genotype-based prediction of CYP450-mediated drug metabolism and the true capacity of an individual to metabolize drugs (phenotype) due to nongenetic factors. Criteria for inclusion were (1) study of the effect of one or more nongenetic factor(s) on CYP450-mediated drug metabolism, (2) availability of relevant CYP450 genotypes resulting in a predicted metabolizer phenotype, and (3) a quantitative assessment of the true CYP450 metabolizer phenotype. The impact of phenoconversion is hypothesized to differ amongst different genotypes. Therefore, studies that solely examined the consequences of comedications for single genotypes were excluded from this review.
2.2. Data Extraction
For the structure of this review, phenoconversion is divided into two topics: assessing the effect of (1) concomitant drugs and other extrinsic factors such as smoking, and (2) patient- and disease-related factors such as comorbidity and age. Phenotype assignment was performed according to the latest guidelines [15,20]. For example, if the original paper used the term extensive metabolizers (EM), subjects were assigned NM. Heterozygous NMs (one wild-type allele and one reduced function) were grouped as IMs.
3. Results
3.1. Study Selection
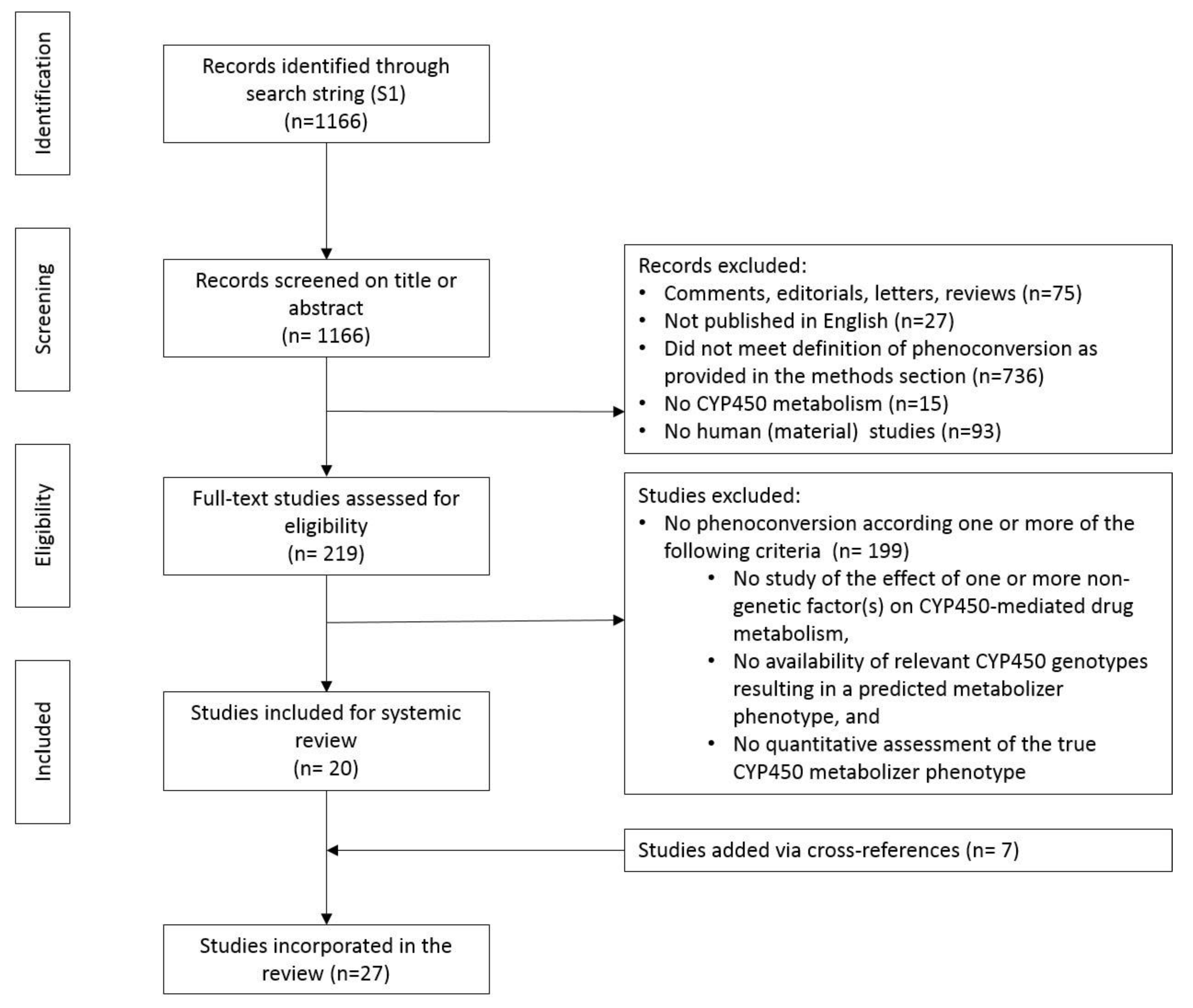
A total of 1166 papers were identified. After screening titles and abstracts, 75 comments, editorials, letters, and reviews and 27 non-English written publications were excluded (Figure 1). A further 736 publications were excluded because they did not meet one or more of our three inclusion criteria (see Methods). Furthermore, 15 publications describing the phenoconversion of non-CYP450 drug metabolism and 93 papers without human (material) data (non-human in-vitro work and modeling studies) were excluded. Of the remaining 219 papers, 199 papers were excluded for lack of relevance after full-text evaluation. Manual checking of references identified seven additional papers. In total, 27 studies were available for analysis.
3.2. Impact of Studies
The 27 studies were divided into 2 groups. Table 1 shows the results for studies investigating the impact of phenoconversion on CYP450 metabolism that results from the use of concomitant drugs and other extrinsic factors. Table 2 shows the results of studies investigating the impact of patient- and disease-related factors on phenoconversion. Two classes of studies could be distinguished: Type 1 studies (n = 15) were specifically designed to study phenoconversion; Type 2 studies (n = 12) were not specifically designed to study phenoconversion and did not categorize patients into PM, IM, NM, RM, or UM phenotype. Typically, these studies showed a change in phenotype, i.e., increased plasma levels as a marker of a decrease in CYP2D6 activity, without assigning a CYP2D6 metabolizer phenotype.
3.3. Phenoconversion of CYP450 by Concomitant Drugs and Other Extrinsic Factors
CYP450 enzymes are involved in the metabolism of 70% to 80% of all marketed drugs [5]. The use of concomitant medication and other extrinsic factors, such as alcohol consumption, smoking, and vitamin D exposure, have all been suggested to influence the occurrence of phenoconversion of CYP450 metabolism and will be described in the next section.
3.3.1. Anticonvulsants
For anticonvulsant drugs, four papers evaluating phenoconversion were identified, three related to comedication-induced phenoconversion, and one related to phenoconversion resulting from alcohol consumption. Stiripentol is a strong inhibitor of CYP1A2, CYP2D6, CYP2C9, CYP2C19, and CYP3A4 [45]. A retrospective study of 28 patients with Dravet syndrome receiving valproate and concomitant stiripentol was undertaken to elucidate the mechanism underlying the increase in serum valproate concentration produced by concomitant stiripentol therapy [21]. It was found that the increase in valproate serum concentration was larger for CYP2C19 NM subjects compared to CYP2C19 PMs, suggesting that CYP2C19 NM subjects are more susceptible to the effects of CYP2C19 inhibition. In a retrospective study of 238 Japanese patients with epilepsy receiving clobazam and concomitant treatment with other antiepileptic drugs, the aim was to identify factors influencing the N-desmethyl-clobazam serum concentration [22]. Clobazam is mainly metabolized by CYP3A4 and partially by CYP2C19 and CYP2B6 into the active metabolite N-desmethyl-clobazam [46]. This metabolite is subsequently converted by CYP2C19 into the inactive metabolite 4′-hydroxynorclobazam. It was shown that concomitant treatment with the CYP450-inducing drugs phenytoin and carbamazepine resulted in a reduced concentration–dose ratio of clobazam and an increased ratio of N-desmethyl-clobazam for CYP2C19 NM and CYP2C19 IM subjects. However, within the CYP2C19 PM group, only the clobazam ratio decreased and not the ratio of N-desmethyl-clobazam, which requires CYP2C19 for further metabolism. The concomitant treatment of zonisamide (a CYP2C19 substrate) and stiripentol (a strong CYP2C19 inhibitor) elevated the concentration–dose ratio of N-desmethyl-clobazam for CYP2C19 NM and CYP2C19 IM subjects. In contrast, the ratio of the CYP2C19 PM group was unchanged, indicating phenoconversion of CYP2C19 NMs and CYP2C19 IMs. In a study using liver microsomes from 114 organ transplants, the donors’ demographic data, drug use, and pathological conditions were explored as potential causes of discrepancies in the genotype-based prediction of CYP2C19 activity [8]. CYP2C19 activity was assessed with mephenytoin and compared with the CYP2C19 genotype. It was found that based on CYP2C19 genetics, the CYP2C19 metabolizer phenotype was overestimated in 47% of the individuals. Chronic alcohol consumption was found to be a factor reducing CYP2C19 activity in 13% of subjects with overestimated CYP2C19 activity [8].
3.3.2. Anticoagulants
One paper evaluating phenoconversion related to comedication in patients taking anticoagulant drugs was identified. In a prospective study of 82 heart surgery patients (CYP2C9*1/*1 and VKORC1 T/T), the influence of the CYP2C19 genotype on the occurrence of bleeding events during treatment with warfarin was investigated [23]. Half of the patients (n = 41) were concomitantly treated with lansoprazole (a CYP2C19 inhibitor) and the other half (n = 41) with rabeprazole (control group). Warfarin dose prior to concomitant treatment with a proton-pump inhibitor was based on their international normalized ratio (INR). The outcome of the study was the number of bleeding events, such as tarry stool, bleeding from colon diverticulum, conjunctival bleeding, or bleeding into the shoulder joint or surgical sites. The study found that during the 2-month follow-up, 24% of patients treated with the CYP2C19 inhibitor lansoprazole experienced bleeding events postsurgery compared to patients treated with rabeprazole (0%), which does not affect CYP2C19. Further examination of the outcomes in lansoprazole-treated patients based on the patients’ predicted phenotype revealed that 40% of the CYP2C19 IM patients experienced a bleeding event. Comparatively, only 12% and 22% of CYP2C19 NM and PM patients, respectively, experienced bleeding events, thus indicating that CYP2C19 inhibition by lansoprazole has a phenotype-mediated effect in patients treated with warfarin.
3.3.3. Antihypertensives
One paper evaluating comedication-related phenoconversion in patients taking antihypertensive drugs was included. In a prospective study of 16 male psychiatric patients treated with thioridazine, the influence of thioridazine withdrawal on the debrisoquine (a CYP2D6 probe drug) MR was evaluated, together with the effect of the CYP2D6 genotype [24]. When treated with thioridazine (150 mg/d or higher), fourteen patients (87.5%) were phenotypical CYP2D6 PM subjects (MR > 12.6). After complete withdrawal of thioridazine in ten patients, only two genotypical CYP2D6 PM patients remained phenotypical CYP2D6 PM. All of the CYP2D6 NM genotype patients returned to CYP2D6 phenotypical NMs after complete withdrawal. At 50 and 100 mg of thioridazine, the proportion of phenotypical CYP2D6 NM subjects decreased to 33% and 29%, respectively. Genotypic CYP2D6 IM patients receiving 50 mg of thioridazine per day were all converted to the CYP2D6 PM metabolizer phenotype, compared to 67% of the genotypic CYP2D6 NM patients. These results show that genotypic CYP2D6 IMs are more susceptible to phenoconversion by thioridazine compared to CYP2D6 NM. Moreover, these results indicate that concomitant use of thioridazine results in phenoconversion with a dose-dependent effect.
3.3.4. Antimuscarinics
One study evaluated phenoconversion during treatment with the antimuscarinic drug tolterodine. In a prospective study of 9 depressed patients, the influence of concomitant treatment with the CYP2D6 inhibitor fluoxetine on the oral clearance of tolterodine was investigated [25]. The authors reported that the oral clearance of tolterodine decreased by 80% in CYP2D6 NM subjects and by 93% in CYP2D6 IM subjects. Even though no formal classification was given of the observed CYP2D6 metabolizer phenotypes, the data suggest that CYP2D6 IMs were more frequently converted to the CYP2D6 PM metabolizer phenotype compared to CYP2D6 NMs.
3.3.5. Antipsychotics
For antipsychotics, four papers evaluating phenoconversion were included: three attributed phenoconversion to comedication, and one investigated smoking-related phenoconversion. In a prospective study of 82 psychiatric patients receiving treatment with one of the CYP2D6 substrates aripiprazole, haloperidol, paliperidone, risperidone or zuclopenthixol, the concomitant use of a CYP2D6 inhibitor was investigated [26]. The study aimed to evaluate the impact of CYP2D6 polymorphisms on antipsychotic concentration accounting for comedication with CYP2D6 inhibitors. Phenoconversion was found to be the limiting factor for predicting an accurate dose based on CYP2D6 genotype. With the exception of paliperidone, patients concomitantly treated with a CYP2D6 inhibitor had increased antipsychotic drug concentrations. Eight out of 82 patients showed a different CYP2D6 metabolizer phenotype from that predicted based on their CYP2D6 genotype. Five genotypically predicted CYP2D6 NM patients were converted to phenotypical CYP2D6 PMs, and one genotypically predicted CYP2D6 IM was a phenotypical CYP2D6 PM. Moreover, 3 genotype-predicted CYP2D6 NMs were phenotypical CYP2D6 IMs. In another prospective study of 93 psychiatric patients treated with the atypical antipsychotic aripiprazole, the effect of concomitant treatment with CYP2D6 inhibitors, aripiprazole plasma concentrations, was evaluated [27]. It was reported that the increase in aripiprazole concentration depended on the CYP2D6 genotype-based prediction of the metabolizer phenotype. Aripiprazole concentrations increased, with 50% for CYP2D6 NM subjects and 20% for CYP2D6 UMs, resulting in an increased risk of moderate and severe side effects.
The interaction between smoking and CYP1A2 is well-established [47]. Diluted smoke condensates bind to and activate the aryl hydrocarbon receptor. Once activated, the aryl hydrocarbon receptor induces the expression of the CYP1A1 and CYP1A2 enzymes [48]. However, the magnitude of this interaction may depend on the CYP1A2 genotype. Consequently, it was hypothesized that smokers with a specific CYP1A2*1F-allele may require higher doses than nonsmokers of drugs metabolized by CYP1A1 and CYP1A2. In a prospective study of 37 patients with schizophrenia or schizoaffective disorder treated with olanzapine, the influence of smoking and CYP1A2 polymorphisms on the disposition of olanzapine and its metabolite 4′-N-desmethylolanzapine in serum and cerebrospinal fluid was investigated [28]. The authors reported a 132% and 107% increase of the 4′-N-desmethylolanzapine/olanzapine ratio in homozygous CYP1A2*1F smokers compared to smokers with other CYP1A2 polymorphisms or nonsmokers that are homozygous carriers of CYP1A2*1F. The increase in the metabolite to parent ratio indicates reduced CYP1A2 enzyme activity in CYP1A2*1F smokers. This study suggests that phenoconversion by smoking is strongly influenced by the CYP1A2 genotype; as a consequence, CYP1A2*1F smokers may require higher doses of olanzapine.
3.3.6. Antitussive and Pain Medication
Two studies evaluated phenoconversion during antitussive and pain medication usage. In a prospective crossover study of 17 healthy volunteers, the effects of concomitant treatment with a moderate CYP2D6 inhibitor (the antidepressant duloxetine) or a strong CYP2D6 inhibitor (paroxetine) on the plasma levels of the CYP2D6 substrates tramadol and dextromethorphan were evaluated [29]. The study aimed to assess the risk of CYP2D6 phenoconversion for CYP2D6 NMs when administered with a moderate or strong CYP2D6 inhibitor. The authors reported the difference in tramadol and dextromethorphan plasma concentrations between homozygous (CYP2D6*1/*1) and heterozygous (CYP2D6*1/*17) NM subjects. It was found that 71% of the heterozygous CYP2D6 NMs and 25% of the homozygous CYP2D6 NMs were converted to phenotypically CYP2D6 IMs by duloxetine. For paroxetine, 94% of the heterozygous CYP2D6 NMs and 56% of the homozygous CYP2D6 NMs were converted to phenotypical CYP2D6 PMs. Nearly all of the heterozygous CYP2D6 NM subjects were phenoconverted with concomitant treatment of paroxetine, while 71% of subjects were phenoconverted with duloxetine. Interestingly, in homozygous CYP2D6 IM subjects, 56% were phenoconverted when treated with paroxetine, but only 25% were phenoconverted when treated with duloxetine. These results show that the strength of the inhibitor (weak/moderate/strong) is important in the influence of phenoconversion, along with genotype. In a follow-up in-vitro study using human liver microsomes, the authors showed that the phenoconversion is likely to be explained through a difference in the amount of functional CYP2D6 enzyme and not by differences in the potency of the CYP2D6 inhibitors for the different genotypes [49]. In a prospective study of 14 Japanese schizophrenia patients, the influence of comedication with neuroleptics metabolized by CYP2D6 was investigated [6]. It was reported that concomitant administration of levomepromazine and biperiden result in higher chlorpromazine levels. Out of the 14 studied patients, 3 CYP2D6 NM patients were converted into CYP2D6 PMs and 2 CYP2D6 NMs into CYP2D6 IMs. The other 9 patients remained CYP2D6 IMs.
3.3.7. Cardiac Drugs
One paper examining phenoconversion in patients taking cardiac drugs was included. In a prospective study, 143 supraventricular tachyarrhythmia patients were concomitantly treated with flecainide and bepridil, a CYP2D6 inhibitor [50]. The objective of the study was to investigate the effects of the CYP2D6 genotype and CYP2D6 inhibitor usage on the serum S-to-R flecainide ratio. Regardless of the genotype status, all 17 concomitant medicated patients had a lower flecainide serum level, resulting in the conversion of CYP2D6 NMs to CYP2D6 IMs or CYP2D6 PMs (CYP2D6 IMs and CYP2D6 PMs were grouped together).
3.3.8. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
One study examined phenoconversion by concomitant medication in patients using NSAIDs. In a prospective crossover study of 22 healthy volunteers, the consequences of 7-day concomitant treatment with flurbiprofen (a CYP2C9 substrate) and fluconazole (a CYP2C9 inhibitor) was investigated [30]. Coadministration of 200 mg fluconazole increased the area under the curve (AUC) of flurbiprofen in predicted CYP2C9 NM (n = 11) to an AUC which is comparable with the AUC of CYP2C9 IM subjects (n = 8), and a dose of 400 mg fluconazole increased the AUC even further compared to CYP2C9 PMs (n = 2). In predicted CYP2C9 IM subjects, both dosages of fluconazole increased the AUC of flurbiprofen, comparable with the AUCs of CYP2C9 PM subjects. These results indicate that the concomitant use of fluconazole can cause phenoconversion in a dose- and genotype-dependent manner.
3.3.9. Proton Pump Inhibitors
A study investigating phenoconversion in patients treated with proton pump inhibitors was included in this review. Fluvoxamine is an inhibitor of CYP1A2, CYP2C19, and CYP3A4. In a prospective study of 18 healthy CYP2C19 genotyped volunteers, the effect of fluvoxamine (inhibitor of CYP1A2, CYP2C19, and CYP3A4) on the pharmacokinetics of lansoprazole (CYP2C19 substrate) was investigated. Subjects received lansoprazole with and without concomitant treatment with fluvoxamine [31]. With concomitant fluvoxamine treatment, CYP2C19 inhibition was reported to be genotype-dependent, with a 2.2- and 1.9-fold decrease for CYP2C19 NM and IM, respectively, compared to administration of lansoprazole alone. This suggests the possible phenoconversion to a lower CYP2C19 metabolizer phenotype (i.e., CYP2C19 IM conversion to IM or PM and IM conversion to PM). There was no significant difference in lansoprazole concentration in CYP2C19 PMs after concomitant fluvoxamine or placebo treatment.
3.3.10. Antiestrogenic Drugs
Two papers investigated phenoconversion in patients using antiestrogenic drugs: one paper due to concomitant medication, and one paper due to vitamin D exposure. In a prospective cohort study, 158 breast cancer patients were treated with tamoxifen and received concomitant treatment with CYP2D6 inhibitors [32]. The study’s aim was to show that concomitant treatment with CYP2D6 inhibitors reduces the endoxifen plasma concentration. Out of the 158 patients, 17 patients used potent CYP2D6 inhibitors (paroxetine and fluoxetine), and 25 patients used weak CYP2D6 inhibitors (sertraline, citalopram, celecoxib, diphenhydramine, and chlorpheniramine). Eighty-five patients not taking any concomitant CYP2D6 medication were used as a control group. With the exception of CYP2D6 UMs, all patients receiving concomitant treatment with a strong CYP2D6 inhibitor were phenoconverted to the CYP2D6 PM metabolizer phenotype. The CYP2D6 UM patients treated with weak or potent CYP2D6 inhibitors showed lower endoxifen plasma concentrations, comparable to the CYP2D6 NM or IM metabolizer phenotype. Concomitant treatment with a weak CYP2D6 inhibitor resulted in patients with the CYP2D6 NM genotype being converted to the IM metabolizer phenotype and IM patients being converted to the PM metabolizer phenotype. This, again, shows that in addition to the genotype, the potency of the inhibitor also plays an important role in the occurrence of phenoconversion.
Vitamin D levels can induce CYP3A4 expression by binding of the vitamin D receptor complexes to proximal promotor elements [51,52]. In a prospective study of 196 breast cancer patients receiving tamoxifen, plasma concentrations of tamoxifen and its metabolite endoxifen were evaluated [33]. The study’s aim was to gain a better understanding of the interpatient variation in endoxifen levels. The suggested induction of CYP3A4 by vitamin D was shown to influence tamoxifen and endoxifen levels. Endoxifen levels were ~20% lower during winter months (January–March) and ~8% higher during summer months (July–September) compared to average levels. Additionally, patients taking vitamin D supplements tended to have higher endoxifen levels compared to patients without supplements. In addition, patients with higher vitamin D levels were more likely to have tamoxifen levels within the therapeutic range.
3.4. Phenoconversion of CYP450 by Patient- and Disease-Related Factors
In addition to the use of extrinsic factors, patient- and disease-related factors such as age, comorbidities, and pregnancy may also affect the relationship between the genotype and the CYP450 metabolizer phenotype. For example, it is known that increased proinflammatory cytokines suppress CYP3A and CYP2C19 activity [53]. A number of diseases, such as infections, are known to show elevated cytokine levels.
3.4.1. Age
One paper evaluating age-related phenoconversion was included. A prospective study that compared the CYP2C19 genotype and pharmacokinetics of omeprazole in young (21–36 years) and elderly (66–85 years) patients found that elderly CYP2C19 NMs show a wider variance in omeprazole pharmacokinetics compared to young CYP2C19 NMs [34]. Importantly, participants were not taking any medication that could affect CYP2C19 metabolism, which excludes the possibility of potential confounding by concomitant medication use in the elderly population. The data showed that 38% of the elderly subjects with the CYP2C19 NM genotype and 42% of the elderly subjects with the CYP2C19 IM genotype were phenotypically CYP2C19 PMs. There were no phenotypical CYP2C19 PM subjects in the genotypical CYP2C19 NM or IM groups among the younger subjects. Moreover, overall plasma levels in elderly CYP2C19 NM and CYP2C19 IM subjects were closer to the CYP2C19 PM metabolizer phenotype compared to younger subjects with CYP2C19 NM or CYP2C19 IM genotypes, demonstrating phenoconversion in the elderly.
3.4.2. Cancer
Three papers evaluating the effect of cancer on the occurrence of phenoconversion were identified. In lung cancer patients, CYP2D6 genotype–phenotype mismatches had already been reported more than 25 years ago [54]. However, these mismatches were not recognized as phenoconversion at that time. The researchers speculated that if the mismatch could be explained by the disease state, the tumor, tumor products, or tumor treatment could modulate the expression (phenoconversion). However, another explanation could be the limitations of the applied genotyping since the used assay only interrogated a limited number of genetic variants. In a prospective study of 16 patients with advanced cancer and a CYP2C19 NM genotype, the relationship between the CYP2C19 genotype and phenotype was studied [35]. Patients received omeprazole and omeprazole, and metabolite levels were measured. It was found that 25% of the patients had an omeprazole hydroxylation index of 1, which indicates a CYP2C19 PM phenotype. Included patients received the same anticancer therapy and did not take CYP2C19 inhibitors, indicating conversion of CYP2C19 NM to CYP2C19 PM due to the cancer disease state. A similar study of 33 patients with advanced cancer aimed to replicate these results [36]. Out of the 33 patients, 30 were genotypically CYP2C19 NM and 3 CYP2C19 IM. Out of the 30 NM, 37% was phenoconverted into the CYP2C19 PM metabolizer phenotype. This effect was not associated with proinflammatory cytokines or growth hormone levels. The authors report a possible association between decreased CYP2C19 activity and low BMI due to cachexia. A prospective study of 25 multiple myeloma patients compared the occurrence of phenoconversion in hematological malignancies and solid tumors [37]. Patients were treated with 200 mg proguanil, and CYP2C19 genotype and proguanil and cycloguanil levels were analyzed. Based on genotyping, no CYP2C19 PMs were predicted for this cohort. However, based on proguanil metabolism, 27% of the genotypically predicted CYP2C19 NMs and 53% of the genotypically predicted CYP2C19 IMs were phenotypically CYP2C19 PMs.
3.4.3. Inflammation
Six studies evaluated phenoconversion as a result of inflammation. In a prospective study of 52 patients with Behcet’s disease and 96 healthy volunteers, the influence of Behcet’s disease and CYP2C9 genotype on the activity of CYP2C9 (phenotypical determined by measuring the metabolic ratio of losartan) was determine [38]. The 31 patients that were genotypically classified as CYP2C9 NM had a mean metabolic ratio that was comparable to the observed metabolic ratio of losartan in CYP2C9 IM healthy volunteers. These results indicate that Behçet’s disease, a systemic inflammatory disorder of the blood vessels, can cause phenoconversion. In a prospective study of 31 patients with hepatitis C virus (HCV)-positive chronic hepatitis or cirrhosis and 30 healthy volunteers, the interaction between chronic liver disease and the CYP2C19 genotype was assessed [39]. Metabolic ratios of omeprazole/5-hydroxy omeprazole were used as the phenotypic test of CYP2C19 activity. In healthy volunteers, metabolic ratios of 0.81, 1.55, and 15.5 were observed in CYP2C19 NM, IM, and PM, respectively. In contrast, patients with chronic liver disease, genotypically classified as CYP2C19 NM, IM, and PM, displayed metabolic ratios of 17.15 (21.1-fold change), 20.02 (12.4-fold change), and 26.04 (1.9-fold change), respectively, which would phenotypically classify them all as CYP2C19 poor metabolizers. These results demonstrate that chronic liver disease resulting from HCV infection can reduce CYP2C19 enzymatic activity and cause phenoconversion in an infection- and genotype-dependent manner. A prospective study of 36 patients treated with intravenous or oral voriconazole aimed to investigate the effect of inflammation on the voriconazole metabolic ratio. It was found that for CYP2C19 UMs, NMs, and IMs, at higher C-reactive protein (CRP) concentrations, the metabolic ratio was decreased, and, consequently, the voriconazole trough concentration was increased [40]. The metabolic ratio was decreased most for CYP2C19 UM subjects and the least for CYP2C19 IMs, suggesting that the degree of phenoconversion is influenced by its genotype. In a study using microsomes from 114 organ transplants described above (see alcohol consumption), the effect of inflammation on phenoconversion was investigated [8]. Diseases with inflammatory processes such as rheumatoid arthritis and gastrointestinal perforation were found to be associated with a reduction of CYP2C19 activity in 47% of the study population. The effect of inflammation on the phenoconversion of CYP2D6 was investigated in a prospective study in 1723 patients with a chronic HCV infection [41]. HCV infection is associated with the presence of liver kidney microsomal type 1 (LKM-1) antibodies, which are directed against the body’s endogenous CYP2D6. It was hypothesized that the LKM-1 antibodies could be a factor influencing the CYP2D6 genotype–phenotype mismatch. When LKM-1-negative patients were compared to LKM-1-positive patients, up to six-fold reduction of CYP2D6 metabolic activity was found in patients with a high level of LKM-1 antibodies (the positive patients). The metabolizer phenotype was explained by genotype in 3 out of the 10 LKM-1-positive subjects. For the others, six CYP2D6 NM subjects were converted to the CYP2D6 IM metabolizer phenotype, and one CYP2D6 NM was converted to the CYP2D6 PM metabolizer phenotype, showing phenoconversion in 70% of the cases. In a prospective study of 63 stable kidney transplant recipients, CYP3A phenoconversion was studied. The CYP3A5 genotype was determined, and immunosuppressant levels were measured [42]. The authors reported that 10 CYP3A5*1 recipients had a lower CYP3A5 activity than expected based on the CYP3A5 genotype, indicating phenoconversion. It was found that higher indoxyl sulfate plasma concentrations were associated with phenoconversion of CYP3A5. While the exact mechanism remains unknown, it is recognized that indoxyl sulfate upregulates the activity of nuclear factor κΒ (the most important inflammatory transcription factor), and nuclear factor κΒ decreases histone 4 acetylation in the CYP3A promotor, which downregulates CYP3A expression [55,56]. Consequently, in patients with the CYP3A5*1 allele and high blood indoxyl sulfate, the dose may need to be adjusted for drugs metabolized by CYP3A [42].
3.4.4. Pregnancy
For pregnancy, two papers assessing phenoconversion were included. In a prospective study of 140 women, the effect of pregnancy on CYP2D6 activity was investigated [43]. To assess CYP2D6 activity, dextromethorphan/dextrorphan metabolic ratios were measured during and after pregnancy. It was reported that 29% and 63% of subjects with a CYP2D6 NM and CYP2D6 IM genotype showed a decreased metabolic ratio, indicating increased CYP2D6 activity. By contrast, CYP2D6 PMs showed an increased metabolic ratio during pregnancy. These observations show that CYP2D6 activity may be induced in CYP2D6 NM and IM but not in CYP2D6 PM subjects during pregnancy. In another prospective study of 74 pregnant women receiving the antidepressant paroxetine, comparable results were reported [44]. CYP2D6 enzymatic activity was increased in genotypic subjects with CYP2D6 NM and UM genotypes and reduced in subjects with a CYP2D6 PM genotype. For CYP2D6 IMs, it was found that the paroxetine plasma concentration is not affected by pregnancy. The authors suggest that the increase of paroxetine plasma levels in genotypic CYP2D6 PMs can be explained by a decreased activity of other CYP450 enzymes involved in paroxetine metabolism. Interestingly, the two studies investigating the effect of pregnancy on phenoconversion reported conflicting results for CYP2D6 IMs and CYP2D6 PMs. A potential explanation could be that the drug studies are not specific probes for CYP2D6 and are metabolized by additional CYP450 enzymes that have reduced activity during pregnancy. For CYP2D6 PMs, the result is an altered metabolic capacity; for CYP2D6 IM subjects, both processes (induced and reduced metabolic capacity) will middle out, resulting in an unaffected plasma concentration [44].
In summary, the phenoconversion of CYP450 due to extrinsic factors has been mostly studied for the use of concomitant medication. In addition, the use of alcohol, smoking, and vitamin D exposure have been described. Out of the 14 studies, six studies formally concluded that phenoconversion occurred. The most frequently described effect was the conversion of the NM metabolizer phenotype to the PM metabolizer phenotype because of the concomitant use of CYP2C19 or CYP2D6 inhibitors. The studies show that inhibition of the different CYP450 enzymes results in lower enzyme activity and, consequently, in higher drug levels of the substrates (Figure 2). By contrast, the use of a CYP450 inducer such as carbamazepine or smoking results in the gain of function of CYP450 enzyme activity (Figure 2). The occurrence of phenoconversion due to patient- and disease-related factors has received very limited attention to date. Only four factors of phenoconversion, age, cancer, inflammation, and pregnancy, have been studied. Nevertheless, this resulted in 12 studies, of which eight reported phenoconversion occurring in ~30% of the patients. The studied disease states included cancer and inflammation, resulting in physiological changes that include, for example, nutrition or higher cytokine levels. These studies provide evidence supporting the hypothesis that higher cytokine levels result in decreased CYP450 activity, resulting in lower drug metabolism and higher drug plasma (Figure 2). The studied patient-related factors are age and pregnancy; age resulted in phenoconversion into a lower metabolizer phenotype due to decreased CYP450 enzyme activity (Figure 2). In conclusion, concomitant medication and distinct patient- and disease-related factors have been shown to modulate the activity of the critical CYP450 enzymes, which resulted in changes in drug metabolism that could not have been predicted from its genotype nor by simply reviewing it as a traditional drug–drug interaction.
4. Discussion
In this systematic review, we assessed studies that investigated phenoconversion by concomitant drugs and other extrinsic factors and patient- and disease-related factors. Out of the 27 identified studies, 10 studies demonstrate phenoconversion for a specific genotype group. Phenoconversion into a lower metabolizer phenotype was reported for concomitant use of CYP450-inhibiting drugs, increasing age, cancer, and inflammation. Phenoconversion into a higher metabolizer phenotype was reported for CYP450 inducers and smoking. Moreover, alcohol consumption, pregnancy, and vitamin D exposure are factors where the study data suggested phenoconversion. The interplay between genetic and nongenetic factors that results in phenoconversion should, therefore, be considered more often.
The use of concomitant medication is frequently reported as a source of phenoconversion. This finding is not unexpected, as the impact of DDIs on drug pharmacokinetics is well established and part of the daily routine of pharmacists. Importantly, several studies included in this review show that the outcome of specific DDIs varies between different genotypes [22,23,27,29,30,31,32,57], a finding that is currently not accounted for in clinical decision-making. As expected, the concomitant administration of strong CYP2D6 or CYP2C19 inhibitors caused phenoconversion in almost all subjects. However, reported data suggest that genotypic IM may be more susceptible to phenoconversion by CYP2D6, CYP2C9, and CYP2C19 inhibitors than PMs, UMs, RMs, or NMs. This finding may be particularly important for the concomitant use of weak or moderate CYP450 inhibitors, which hypothetically will have the strongest effect on phenoconversion in IM subjects and warrant further investigations.
Another recognized source of phenoconversion of CYP450 drug metabolism are comorbidities, including cancer. Several studies included in this review established that the effect of specific drug–disease interactions on CYP450 metabolism varies between different CYP450 genotypes [35,36,37,41,42]. In contrast to phenoconversion resulting from external factors, the effects of comorbidities are not yet taken into account in the management of drug treatment. Moreover, the exact mechanisms underlying phenoconversion induced by disease states such as cancer and inflammation remain to be elucidated. Liver diseases have also been suggested as a source of phenoconversion. However, while a number of studies did show changes in plasma levels due to liver diseases, no adequate genotype assessments were available in those studies [58,59,60]. Therefore, for disease-induced phenoconversion, further studies are also necessary to help to integrate pharmacogenetic information for personalizing the control of drug–disease interactions.
Our systematic search identified only 27 eligible studies investigating phenoconversion. Most of the identified studies were not specifically designed to study phenoconversion and included retrospective analyses with small sample sizes and limited genotyping. While quantitative assessment of the true CYP450 metabolizer phenotype was performed, no formal metabolizer phenotype was assigned. The assessed studies reported large numbers of genotype–phenotype discrepancies. Nevertheless, the impact on the effectiveness and toxicity remains mostly unknown. To improve our understanding of phenoconversion studies with comprehensive CYP450 genotyping, a quantitative assessment of the true CYP450 metabolizer phenotype and, ideally, a well-defined drug–response phenotype are required.
The studies included in our review mostly consider drugs that are metabolized by 1-2 CYP450 enzymes. Moreover, the effect of a single concomitant drug or other factor is investigated. However, in reality, many drugs have a complex metabolism, and patients suffer from multiple morbidities and are treated with multiple drugs simultaneously [22,61]. A recent study of patients treated with clozapine investigated the use of genotype- and comedication-corrected scores to predict the impact on clozapine exposure [62]. It was shown that by using the scores, the number of CYP1A2 UMs increased by 1.1-fold, CYP2D6 IMs and PMs by 1.8-fold, and CYP2C19 IM by 1.7-fold, respectively. In the future, this type of model that combines genetic information with information on comorbidities, concomitant drug treatment, and other external factors may help to improve our capability to predict the outcome of drug treatment.
In this review, we have focused on clinical studies investigating phenoconversion. However, other approaches, including the use of in-vitro or in-silico models, may be feasible. For in-vitro studies, this requires model(s) in which the impact of the phenoconversion factor can simultaneously be assessed for different genotypes. This can be achieved either via liver models with endogenous pharmacogenetic variation, such as primary human hepatocytes (PHHs), liver microsomes, or induced pluripotent stem cells (iPSCs) [63,64,65,66], or through hepatocytes in which pharmacogenetic variants are exogenously introduced via CRISPR [67]. Secondly, phenoconversion for some conditions (e.g., inflammation) may only develop after prolonged or repeated exposure, which requires in-vitro models, such as 3D liver spheroids, in which the long-term consequences of these factors for drug metabolism can be adequately studied [68,69]. In-silico models, such as physiologically based pharmacokinetic (PBPK) models, have also been suggested as an excellent approach to study phenoconversion [70,71,72,73,74,75]. These models are particularly suitable to investigate the effects of complex interactions between multiple drugs and disease states.
5. Conclusions
Phenoconversion of CYP450 metabolism is caused by both extrinsic factors, such as the use of concomitant drugs, as well as patient- and disease-related factors. The mechanism(s) behind and extent to which CYP450 metabolism is affected by these factors remain largely unexplored. If studied more comprehensively, accounting for phenoconversion may help to improve our ability to predict the individual CYP450 metabolism and personalize drug treatment.
Author Contributions
Wrote the manuscript: S.D.K., M.L.M., H.-J.G., and J.J.S. Literature search and data extraction: SK. Data interpretation: S.D.K., M.L.M., H.-J.G., and J.J.S. All authors have read and agreed to the published version of the manuscript.
Funding
The collaboration project is cofunded by the PPP Allowance made available by Health~Holland, Top Sector Life Sciences & Health, to stimulate public–private partnerships.
Acknowledgments
We are particularly grateful for the assistance on search terms given by Jan Schoones of the Walaeus library (LUMC).
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A. Search Strategy
((“phenoconversion”[tw] OR phenoconver*[tw] OR “pheno conversion”[tw] OR ((“extensive metabolizer”[tw] OR “extensive metabolizers”[tw] OR “extensive metaboliser”[tw] OR “extensive metabolisers”[tw]) OR (“poor metabolizer”[tw] OR “poor metabolizers”[tw] OR “poor metaboliser”[tw] OR “poor metabolisers”[tw])) OR (“intermediate metabolizer”[tw] OR “intermediate metabolizers”[tw] OR “intermediate metaboliser”[tw] OR “intermediate metabolisers”[tw])) OR (“ultra-rapid metabolizer”[tw] OR “ultra-rapid metabolizers”[tw] OR “ultra-rapid metaboliser”[tw] OR “ultra-rapid metabolisers”[tw])) OR (“rapid metabolizer”[tw] OR “rapid metabolizers”[tw] OR “rapid metaboliser”[tw] OR “rapid metabolisers”[tw]))OR ((genotyp*[tw] AND “extensive”[tw] AND metaboli*[tw]) AND (phenotyp*[tw] AND “poor”[tw] AND metaboli*[tw]))) AND (“Drug Therapy, Combination”[Mesh] OR “Combination Chemotherapy”[tw] OR “Drug Polytherapy”[tw] OR “Drug Polytherapies”[tw] OR “Combination Chemotherapies”[tw] OR “Combination Drug Therapy”[tw] OR “Combination Drug Therapies”[tw] OR “Polychemotherapy”[tw] OR Polychemotherap*[tw] OR “Poly-chemotherapy”[tw] OR Poly-chemotherap*[tw] OR comedicat*[tw] OR co-medicat*[tw] OR OR “Comorbidity”[Mesh] OR “Comorbidity”[tw] OR Comorbid*[tw] OR “Co-morbidity”[tw] OR Co-morbid*[tw] OR “Multimorbidity”[tw] OR Multimorbid*[tw] OR “Multi-morbidity”[tw] OR Multi-morbid*[tw] OR “Cytokines”[mesh] OR “cytokines”[tw] OR “cytokine”[tw] OR “Interleukin-4”[mesh] OR “Interleukin-6”[mesh] OR “Interleukin-13”[mesh] OR “Interleukin-4”[tw] OR “Interleukin-6”[tw] OR “Interleukin-13”[tw] OR interleukin*[tw] OR “Liver Diseases”[Mesh] OR “Liver Diseases”[tw] OR “Liver Disease”[tw] OR “liver”[tw] OR “hepatic”[tw] OR “Nervous System”[Mesh] OR “Nervous System”[tw] OR “Nervous System Diseases”[Mesh] OR “Transplantation”[Mesh] OR “transplantation”[Subheading] OR transplant*[tw] OR “Inflammation”[Mesh] OR “ Inflammation “[Subheading] OR Inflam*[tw] OR “Cardiac Diseases”[Mesh] OR “Cardiac Diseases”[tw] OR “Cardiac Disease”[tw] OR “hart”[tw] OR “cardiac”[tw] ] OR “Cancer”[Mesh] OR “Cancer”[Subheading] OR Cancer*[tw] OR “Diet”[Mesh] OR “Diets”[tw] OR “Diet”[tw] OR “Fasting”[Mesh] OR “Fasting”[tw] OR “Smoking”[Mesh] OR “Smoking”[tw] OR “Alcohol Drinking”[Mesh] OR “Alcohol Drinking”[tw] OR “Alcoholism”[Mesh] OR “Alcoholism”[tw] OR “Pregnancy”[mesh] OR pregnan*[tw]) AND (“Cytochrome P-450 Enzyme System”[Mesh] OR “Cytochrome P-450”[tw] OR “Cytochrome P450”[tw] OR “25-Hydroxyvitamin D3 1-alpha-Hydroxylase”[tw] OR “Aniline Hydroxylase”[tw] OR “Aromatase”[tw] OR “Aryl Hydrocarbon Hydroxylases”[tw] OR “Benzopyrene Hydroxylase”[tw] OR “Camphor 5-Monooxygenase”[tw] OR “Cholestanetriol 26-Monooxygenase”[tw] OR “Cholesterol 24-Hydroxylase”[tw] OR “Cholesterol 7-alpha-Hydroxylase”[tw] OR “Cholesterol Side-Chain Cleavage Enzyme”[tw] OR “CYP11B2”[tw] OR “CYP1A1”[tw] OR “CYP1A2”[tw] OR “CYP1B1”[tw] OR “CYP2A6”[tw] OR “CYP2B1”[tw] OR “CYP2B6”[tw] OR “CYP2C19”[tw] OR “CYP2C9”[tw] OR “CYP2C8”[tw] OR “CYP2D6”[tw] OR “CYP2E1”[tw] OR “CYP3A”[tw] OR “CYP4A”[tw] OR “Limonene Hydroxylases”[tw] OR “Retinoic Acid 4-Hydroxylase”[tw] OR “Steroid 11-beta-Hydroxylase”[tw] OR “Steroid 12-alpha-Hydroxylase”[tw] OR “Steroid 16-alpha-Hydroxylase”[tw] OR “Steroid 17-alpha-Hydroxylase”[tw] OR “Steroid 21-Hydroxylase”[tw] OR “Steroid Hydroxylases”[tw] OR “Sterol 14-Demethylase”[tw] OR “Trans-Cinnamate 4-Monooxygenase”[tw] OR “Vitamin D3 24-Hydroxylase”[tw]))
References
- Abbasi, J. Getting Pharmacogenomics into the Clinic. JAMA 2016, 316, 1533–1535. [Google Scholar] [CrossRef] [PubMed]
- Roden, D.M.; McLeod, H.L.; Relling, M.V.; Williams, M.S.; Mensah, G.A.; Peterson, J.F.; Van Driest, S.L. Pharmacogenomics. Lancet 2019, 394, 521–532. [Google Scholar] [CrossRef]
- Relling, M.V.; Klein, T.E. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin. Pharm. Ther. 2011, 89, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Swen, J.J.; Nijenhuis, M.; de Boer, A.; Grandia, L.; Maitland-van der Zee, A.H.; Mulder, H.; Rongen, G.A.; van Schaik, R.H.; Schalekamp, T.; Touw, D.J.; et al. Pharmacogenetics: From bench to byte—An update of guidelines. Clin. Pharm. Ther. 2011, 89, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharm. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Ieiri, I.; Yamada, S.; Seto, K.; Morita, T.; Kaneda, T.; Mamiya, K.; Tashiro, N.; Higuchi, S.; Otsubo, K. A CYP2D6 phenotype-genotype mismatch in Japanese psychiatric patients. Pharmacopsychiatry 2003, 36, 192–196. [Google Scholar] [CrossRef]
- Rost, K.L.; Brockmoller, J.; Esdorn, F.; Roots, I. Phenocopies of poor metabolizers of omeprazole caused by liver disease and drug treatment. J. Hepatol. 1995, 23, 268–277. [Google Scholar] [CrossRef]
- Kiss, A.F.; Vasko, D.; Deri, M.T.; Toth, K.; Monostory, K. Combination of CYP2C19 genotype with non-genetic factors evoking phenoconversion improves phenotype prediction. Pharm. Rep. 2018, 70, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.R.; Gaedigk, A.; LLerina, A.; Eichelbaum, M.; Stingl, J.; Smith, R.L. CYP450 genotype and pharmacogenetic association studies: A critical appraisal. Pharmacogenomics 2016, 17, 259–275. [Google Scholar] [CrossRef]
- Karle, J.; Bolbrinker, J.; Vogl, S.; Kreutz, R.; Denkert, C.; Eucker, J.; Wischnewsky, M.; Possinger, K.; Regierer, A.C. Influence of CYP2D6-genotype on tamoxifen efficacy in advanced breast cancer. Breast Cancer Res. Treat. 2013, 139, 553–560. [Google Scholar] [CrossRef] [Green Version]
- D’Empaire, I.; Guico-Pabia, C.J.; Preskorn, S.H. Antidepressant treatment and altered CYP2D6 activity: Are pharmacokinetic variations clinically relevant? J. Psychiatr. Pract. 2011, 17, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.; Zanger, U. Pharmacogenomics of Cytochrome P450 3A4: Recent Progress Toward the “Missing Heritability” Problem. Front. Genet. 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, R.R.; Smith, R.L. Addressing phenoconversion: The Achilles’ heel of personalized medicine. Br. J. Clin. Pharmacol. 2015, 79, 222–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, R.R.; Smith, R.L. Inflammation-induced phenoconversion of polymorphic drug metabolizing enzymes: Hypothesis with implications for personalized medicine. Drug Metab. Dispos. 2015, 43, 400–410. [Google Scholar] [CrossRef] [Green Version]
- Caudle, K.E.; Sangkuhl, K.; Whirl-Carrillo, M.; Swen, J.J.; Haidar, C.E.; Klein, T.E.; Gammal, R.S.; Relling, M.V.; Scott, S.A.; Hertz, D.L.; et al. Standardizing CYP2D6 Genotype to Phenotype Translation: Consensus Recommendations from the Clinical Pharmacogenetics Implementation Consortium and Dutch Pharmacogenetics Working Group. Clin. Transl. Sci. 2020, 13, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Flockhart, D. Drug Interactions: Cytochrome P450 Drug Interaction Table; Indiana University School of Medicine: Indianapolis, IN, USA, 2007. [Google Scholar]
- Birdwell, K.A.; Decker, B.; Barbarino, J.M.; Peterson, J.F.; Stein, C.M.; Sadee, W.; Wang, D.; Vinks, A.A.; He, Y.; Swen, J.J.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for CYP3A5 Genotype and Tacrolimus Dosing. Clin. Pharm. Ther. 2015, 98, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Counsell, C. Formulating Questions and Locating Primary Studies for Inclusion in Systematic Reviews. Ann. Intern. Med. 1997, 127, 380–387. [Google Scholar] [CrossRef]
- Moher, D.; Shamseer, L.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A.; Group, P.-P. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst. Rev. 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Bank, P.C.D.; Caudle, K.E.; Swen, J.J.; Gammal, R.S.; Whirl-Carrillo, M.; Klein, T.E.; Relling, M.V.; Guchelaar, H.J. Comparison of the Guidelines of the Clinical Pharmacogenetics Implementation Consortium and the Dutch Pharmacogenetics Working Group. Clin. Pharmacol. Ther. 2018, 103, 599–618. [Google Scholar] [CrossRef]
- Jogamoto, T.; Yamamoto, Y.; Fukuda, M.; Suzuki, Y.; Imai, K.; Takahashi, Y.; Inoue, Y.; Ohtsuka, Y. Add-on stiripentol elevates serum valproate levels in patients with or without concomitant topiramate therapy. Epilepsy Res. 2017, 130, 7–12. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Takahashi, Y.; Imai, K.; Miyakawa, K.; Nishimura, S.; Kasai, R.; Ikeda, H.; Takayama, R.; Mogami, Y.; Yamaguchi, T.; et al. Influence of CYP2C19 polymorphism and concomitant antiepileptic drugs on serum clobazam and N-desmethyl clobazam concentrations in patients with epilepsy. Ther. Drug Monit. 2013, 35, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Hata, M.; Shiono, M.; Akiyama, K.; Sezai, A.; Wakui, S.; Kimura, H.; Sekino, H. Incidence of drug interaction when using proton pump inhibitor and warfarin according to cytochrome P450 2C19 (CYP2C19) genotype in Japanese. Thorac. Cardiovasc. Surg. 2015, 63, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Llerena, A.; Berecz, R.; de la Rubia, A.; Fernández-Salguero, P.; Dorado, P. Effect of Thioridazine Dosage on the Debrisoquine Hydroxylation Phenotype in Psychiatric Patients with Different CYP2D6 Genotypes. Ther. Drug Monit. 2001, 23. [Google Scholar] [CrossRef]
- Brynne, N.; Svanstrom, C.; Aberg-Wistedt, A.; Hallen, B.; Bertilsson, L. Fluoxetine inhibits the metabolism of tolterodine-pharmacokinetic implications and proposed clinical relevance. Br. J. Clin. Pharmacol. 1999, 48, 553–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisbeth, P.; Vincent, H.; Kristof, M.; Bernard, S.; Manuel, M.; Hugo, N. Genotype and co-medication dependent CYP2D6 metabolic activity: Effects on serum concentrations of aripiprazole, haloperidol, risperidone, paliperidone and zuclopenthixol. Eur. J. Clin. Pharmacol. 2016, 72, 175–184. [Google Scholar] [CrossRef]
- Kiss, A.; Menus, A.; Toth, K.; Deri, M.; Sirok, D.; Gabri, E.; Belic, A.; Csukly, G.; Bitter, I.; Monostory, K. Phenoconversion of CYP2D6 by inhibitors modifies aripiprazole exposure. Eur. Arch. Psychiatry Clin. Neurosci. 2019. [Google Scholar] [CrossRef] [Green Version]
- Skogh, E.; Sjodin, I.; Josefsson, M.; Dahl, M.L. High correlation between serum and cerebrospinal fluid olanzapine concentrations in patients with schizophrenia or schizoaffective disorder medicating with oral olanzapine as the only antipsychotic drug. J. Clin. Psychopharmacol. 2011, 31, 4–9. [Google Scholar] [CrossRef]
- Storelli, F.; Matthey, A.; Lenglet, S.; Thomas, A.; Desmeules, J.; Daali, Y. Impact of CYP2D6 Functional Allelic Variations on Phenoconversion and Drug-Drug Interactions. Clin. Pharmacol. Ther. 2018, 104, 148–157. [Google Scholar] [CrossRef]
- Kumar, V.; Brundage, R.C.; Oetting, W.S.; Leppik, I.E.; Tracy, T.S. Differential Genotype Dependent Inhibition of CYP2C9 in Humans. Drug Metab. Dispos. 2008, 36, 1242–1248. [Google Scholar] [CrossRef] [Green Version]
- Miura, M.; Tada, H.; Yasui-Furukori, N.; Uno, T.; Sugawara, K.; Tateishi, T.; Suzuki, T. Enantioselective disposition of lansoprazole in relation to CYP2C19 genotypes in the presence of fluvoxamine. Br. J. Clin. Pharmacol. 2005, 60, 61–68. [Google Scholar] [CrossRef]
- Borges, S.; Desta, Z.; Li, L.; Skaar, T.C.; Ward, B.A.; Nguyen, A.; Jin, Y.; Storniolo, A.M.; Nikoloff, D.M.; Wu, L.; et al. Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism: Implication for optimization of breast cancer treatment. Clin Pharm. Ther. 2006, 80, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Teft, W.A.; Gong, I.Y.; Dingle, B.; Potvin, K.; Younus, J.; Vandenberg, T.A.; Brackstone, M.; Perera, F.E.; Choi, Y.H.; Zou, G.; et al. CYP3A4 and seasonal variation in vitamin D status in addition to CYP2D6 contribute to therapeutic endoxifen level during tamoxifen therapy. Breast Cancer Res. Treat. 2013, 139, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, Y.; Yasui-Furukori, N.; Takahata, T.; Sasaki, M.; Tateishi, T. The effect of aging on the relationship between the cytochrome P450 2C19 genotype and omeprazole pharmacokinetics. Clin. Pharmacokinet. 2005, 44, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.L.; Bhargava, P.; Cherrouk, I.; Marshall, J.L.; Flockhart, D.A.; Wainer, I.W. A discordance of the cytochrome P450 2C19 genotype and phenotype in patients with advanced cancer. Br. J. Clin. Pharmacol. 2000, 49, 485–488. [Google Scholar] [CrossRef] [Green Version]
- Helsby, N.A.; Lo, W.Y.; Sharples, K.; Riley, G.; Murray, M.; Spells, K.; Dzhelai, M.; Simpson, A.; Findlay, M. CYP2C19 pharmacogenetics in advanced cancer: Compromised function independent of genotype. Br. J. Cancer 2008, 99, 1251–1255. [Google Scholar] [CrossRef] [Green Version]
- Burns, K.E.; Goldthorpe, M.A.; Porteus, F.; Browett, P.; Helsby, N.A. CYP2C19 genotype-phenotype discordance in patients with multiple myeloma leads to an acquired loss of drug-metabolising activity. Cancer Chemother. Pharmacol. 2014, 73, 651–655. [Google Scholar] [CrossRef]
- Goktaş, M.T.; Hatta, F.; Karaca, O.; Kalkisim, S.; Kilic, L.; Akdogan, A.; Babaoglu, M.O.; Bozkurt, A.; Helldén, A.; Bertilsson, L.; et al. Lower CYP2C9 activity in Turkish patients with Behçet’s disease compared to healthy subjects: A down-regulation due to inflammation? Eur. J. Clin. Pharm. 2015, 71, 1223–1228. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, A.; Murakami, S.; Akizuki, S.; Mochizuki, J.; Echizen, H.; Takagi, I. In vivo metabolic activity of CYP2C19 and CYP3A in relation to CYP2C19 genetic polymorphism in chronic liver disease. J. Clin. Pharmacol. 2005, 45, 1221–1229. [Google Scholar] [CrossRef]
- Veringa, A.; Ter Avest, M.; Span, L.F.; van den Heuvel, E.R.; Touw, D.J.; Zijlstra, J.G.; Kosterink, J.G.; van der Werf, T.S.; Alffenaar, J.C. Voriconazole metabolism is influenced by severe inflammation: A prospective study. J. Antimicrob. Chemother. 2017, 72, 261–267. [Google Scholar] [CrossRef]
- Girardin, F.; Daali, Y.; Gex-Fabry, M.; Rebsamen, M.; Roux-Lombard, P.; Cerny, A.; Bihl, F.; Binek, J.; Moradpour, D.; Negro, F.; et al. Liver kidney microsomal type 1 antibodies reduce the CYP2D6 activity in patients with chronic hepatitis C virus infection. J. Viral Hepat. 2012, 19, 568–573. [Google Scholar] [CrossRef]
- Suzuki, Y.; Muraya, N.; Fujioka, T.; Sato, F.; Tanaka, R.; Matsumoto, K.; Sato, Y.; Ohno, K.; Mimata, H.; Kishino, S.; et al. Factors involved in phenoconversion of CYP3A using 4beta-hydroxycholesterol in stable kidney transplant recipients. Pharmacol. Rep. Pract. 2019, 71, 276–281. [Google Scholar] [CrossRef]
- Wadelius, M.; Darj, E.; Frenne, G.; Rane, A. Induction of CYP2D6 in pregnancy. Clin. Pharmacol. Ther. 1997, 62, 400–407. [Google Scholar] [CrossRef]
- Ververs, F.F.; Voorbij, H.A.; Zwarts, P.; Belitser, S.V.; Egberts, T.C.; Visser, G.H.; Schobben, A.F. Effect of cytochrome P450 2D6 genotype on maternal paroxetine plasma concentrations during pregnancy. Clin. Pharmacokinet. 2009, 48, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Tran, A.; Rey, E.; Pons, G.; Rousseau, M.; d’Athis, P.; Olive, G.; Mather, G.G.; Bishop, F.E.; Wurden, C.J.; Labroo, R.; et al. Influence of stiripentol on cytochrome P450-mediated metabolic pathways in humans: In vitro and in vivo comparison and calculation of in vivo inhibition constants. Clin. Pharmacol. Ther. 1997, 62, 490–504. [Google Scholar] [CrossRef]
- Giraud, C.; Tran, A.; Rey, E.; Vincent, J.; Tréluyer, J.M.; Pons, G. In vitro characterization of clobazam metabolism by recombinant cytochrome P450 enzymes: Importance of CYP2C19. Drug Metab. Dispos. 2004, 32, 1279–1286. [Google Scholar] [CrossRef] [Green Version]
- Kroon, L. Drug interactions with smoking. Am. J. Health Syst. Pharm. 2007, 64, 1917–1921. [Google Scholar] [CrossRef]
- Meek, M.D.; Finch, G.L. Diluted Mainstream Cigarette Smoke Condensates Activate Estrogen Receptor and Aryl Hydrocarbon Receptor-Mediated Gene Transcription. Environ. Res. 1999, 80, 9–17. [Google Scholar] [CrossRef]
- Storelli, F.; Desmeules, J.; Daali, Y. Genotype-sensitive reversible and time-dependent CYP2D6 inhibition in human liver microsomes. Basic Clin. Pharmacol. Toxicol. 2019, 124, 170–180. [Google Scholar] [CrossRef]
- Doki, K.; Sekiguchi, Y.; Kuga, K.; Aonuma, K.; Homma, M. Serum flecainide S/R ratio reflects the CYP2D6 genotype and changes in CYP2D6 activity. Drug Metab. Pharmacokinet. 2015, 30, 257–262. [Google Scholar] [CrossRef]
- Schmiedlin-Ren, P.; Thummel, K.E.; Fisher, J.M.; Paine, M.F.; Lown, K.S.; Watkins, P.B. Expression of enzymatically active CYP3A4 by Caco-2 cells grown on extracellular matrix-coated permeable supports in the presence of 1alpha,25-dihydroxyvitamin D3. Mol. Pharm. 1997, 51, 741–754. [Google Scholar] [CrossRef] [Green Version]
- Thummel, K.E.; Brimer, C.; Yasuda, K.; Thottassery, J.; Senn, T.; Lin, Y.; Ishizuka, H.; Kharasch, E.; Schuetz, J.; Schuetz, E. Transcriptional control of intestinal cytochrome P-4503A by 1alpha,25-dihydroxy vitamin D3. Mol. Pharm. 2001, 60, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Lanchote, V.L.; Almeida, R.; Barral, A.; Barral-Netto, M.; Marques, M.P.; Moraes, N.V.; da Silva, A.M.; Souza, T.M.; Suarez-Kurtz, G. Impact of visceral leishmaniasis and curative chemotherapy on cytochrome P450 activity in Brazilian patients. Br. J. Clin. Pharm. 2015, 80, 1160–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, N.E.; Shields, P.G.; Landi, M.T.; Shaw, G.L.; Tucker, M.A.; Hoover, R.; Sugimura, H.; Weston, A.; Harris, C.C. The debrisoquine metabolic phenotype and DNA-based assays: Implications of misclassification for the association of lung cancer and the debrisoquine metabolic phenotype. Environ. Health Perspect. 1992, 98, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Velenosi, T.J.; Feere, D.A.; Sohi, G.; Hardy, D.B.; Urquhart, B.L. Decreased nuclear receptor activity and epigenetic modulation associates with down-regulation of hepatic drug-metabolizing enzymes in chronic kidney disease. FASEB J. 2014, 28, 5388–5397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Gao, Z.; Wang, L.; Gao, Y. Upregulation of nuclear factor-κB activity mediates CYP24 expression and reactive oxygen species production in indoxyl sulfate-induced chronic kidney disease. Nephrology 2016, 21, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Takahashi, Y.; Imai, K.; Mogami, Y.; Matsuda, K.; Nakai, M.; Kagawa, Y.; Inoue, Y. Interaction between sulthiame and clobazam: Sulthiame inhibits the metabolism of clobazam, possibly via an action on CYP2C19. Epilepsy Behav. 2014, 34, 124–126. [Google Scholar] [CrossRef]
- Frye, R.F.; Zgheib, N.K.; Matzke, G.R.; Chaves-Gnecco, D.; Rabinovitz, M.; Shaikh, O.S.; Branch, R.A. Liver disease selectively modulates cytochrome P450–mediated metabolism. Clin. Pharmacol. Ther. 2006, 80, 235–245. [Google Scholar] [CrossRef]
- Pageaux, G.P.; Micallef, J.; Nataf, M.B.; Levron, J.C.; Lacarelle, B.; Le Moing, J.P.; Bouhours, P.; Blin, O. Pharmacokinetics of sabeluzole and dextromethorphan oxidation capacity in patients with severe hepatic dysfunction and healthy volunteers. Br. J. Clin. Pharmacol. 2001, 51, 164–168. [Google Scholar] [CrossRef]
- Shao, J.G.; Jiang, W.; Li, K.Q.; Lu, J.R.; Sun, Y.Y. Blood concentration of pantoprazole sodium is significantly high in hepatogenic peptic ulcer patients, especially those with a poor CYP2C19 metabolism. J. Dig. Dis. 2009, 10, 55–60. [Google Scholar] [CrossRef]
- Okubo, M.; Murayama, N.; Miura, J.; Chiba, Y.; Yamazaki, H. Effects of cytochrome P450 2D6 and 3A5 genotypes and possible coadministered medicines on the metabolic clearance of antidepressant mirtazapine in Japanese patients. Biochem. Pharmacol. 2015, 93, 104–109. [Google Scholar] [CrossRef]
- Lesche, D.; Mostafa, S.; Everall, I.; Pantelis, C.; Bousman, C.A. Impact of CYP1A2, CYP2C19, and CYP2D6 genotype- and phenoconversion-predicted enzyme activity on clozapine exposure and symptom severity. Pharm. J. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, L.B.; Sorensen, R.N.; Miners, J.O.; Somogyi, A.A.; Grgurinovich, N.; Birkett, D.J. Polymorphic hydroxylation of perhexiline in vitro. Br. J. Clin. Pharmacol. 2003, 55, 635–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbett, J.L.; Duncan, S.A. iPSC-Derived Hepatocytes as a Platform for Disease Modeling and Drug Discovery. Front. Med. 2019, 6, 265. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, J.P.; Hyland, R.; Youdim, K. Minimizing polymorphic metabolism in drug discovery: Evaluation of the utility of in vitro methods for predicting pharmacokinetic consequences associated with CYP2D6 metabolism. Drug Metab. Dispos. Biol. Fate Chem. 2006, 34, 1516–1522. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, M.; Oguchi, A.; Murakawa, Y. Genomic Instability of iPSCs and Challenges in Their Clinical Applications. Adv. Exp. Med. Biol. 2019, 1201, 23–47. [Google Scholar] [CrossRef]
- Dorr, C.R.; Remmel, R.P.; Muthusamy, A.; Fisher, J.; Moriarity, B.S.; Yasuda, K.; Wu, B.; Guan, W.; Schuetz, E.G.; Oetting, W.S.; et al. CRISPR/Cas9 Genetic Modification of CYP3A5 *3 in HuH-7 Human Hepatocyte Cell Line Leads to Cell Lines with Increased Midazolam and Tacrolimus Metabolism. Drug Metab. Dispos. 2017, 45, 957–965. [Google Scholar] [CrossRef]
- Hendriks, D.F.G.; Vorrink, S.U.; Smutny, T.; Sim, S.C.; Nordling, Å.; Ullah, S.; Kumondai, M.; Jones, B.C.; Johansson, I.; Andersson, T.B.; et al. Clinically Relevant Cytochrome P450 3A4 Induction Mechanisms and Drug Screening in Three-Dimensional Spheroid Cultures of Primary Human Hepatocytes. Clin. Pharmacol. Ther. 2020. [Google Scholar] [CrossRef]
- Vorrink, S.U.; Ullah, S.; Schmidt, S.; Nandania, J.; Velagapudi, V.; Beck, O.; Ingelman-Sundberg, M.; Lauschke, V.M. Endogenous and xenobiotic metabolic stability of primary human hepatocytes in long-term 3D spheroid cultures revealed by a combination of targeted and untargeted metabolomics. FASEB J. 2017, 31, 2696–2708. [Google Scholar] [CrossRef] [Green Version]
- Dickschen, K.; Willmann, S.; Thelen, K.; Lippert, J.; Hempel, G.; Eissing, T. Physiologically Based Pharmacokinetic Modeling of Tamoxifen and its Metabolites in Women of Different CYP2D6 Phenotypes Provides New Insight into the Tamoxifen Mass Balance. Front. Pharmacol. 2012, 3, 92. [Google Scholar] [CrossRef] [Green Version]
- Andreu, F.; Colom, H.; Elens, L.; van Gelder, T.; van Schaik, R.H.N.; Hesselink, D.A.; Bestard, O.; Torras, J.; Cruzado, J.M.; Grinyo, J.M.; et al. A New CYP3A5*3 and CYP3A4*22 Cluster Influencing Tacrolimus Target Concentrations: A Population Approach. Clin. Pharmacokinet. 2017, 56, 963–975. [Google Scholar] [CrossRef]
- Woillard, J.B.; Mourad, M.; Neely, M.; Capron, A.; van Schaik, R.H.; van Gelder, T.; Lloberas, N.; Hesselink, D.A.; Marquet, P.; Haufroid, V.; et al. Tacrolimus Updated Guidelines through popPK Modeling: How to Benefit More from CYP3A Pre-emptive Genotyping Prior to Kidney Transplantation. Front. Pharmacol. 2017, 8, 358. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, D.; Tang, W.; Zhou, W.; Al-Huniti, N.; Masson, E. Physiologically Based Pharmacokinetic Modeling to Evaluate the Systemic Exposure of Gefitinib in CYP2D6 Ultrarapid Metabolizers and Extensive Metabolizers. J. Clin. Pharmacol. 2018, 58, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Moj, D.; Britz, H.; Burhenne, J.; Stewart, C.F.; Egerer, G.; Haefeli, W.E.; Lehr, T. A physiologically based pharmacokinetic and pharmacodynamic (PBPK/PD) model of the histone deacetylase (HDAC) inhibitor vorinostat for pediatric and adult patients and its application for dose specification. Cancer Chemother. Pharmacol. 2017, 80, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Titze, M.I.; Schaaf, O.; Hofmann, M.H.; Sanderson, M.P.; Zahn, S.K.; Quant, J.; Lehr, T. A comprehensive pharmacokinetic/pharmacodynamics analysis of the novel IGF1R/INSR inhibitor BI 893923 applying in vitro, in vivo and in silico modeling techniques. Cancer Chemother. Pharmacol. 2016, 77, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Study flow diagram of the retrieval and review process (Appendix A).
Figure 1.
Study flow diagram of the retrieval and review process (Appendix A).

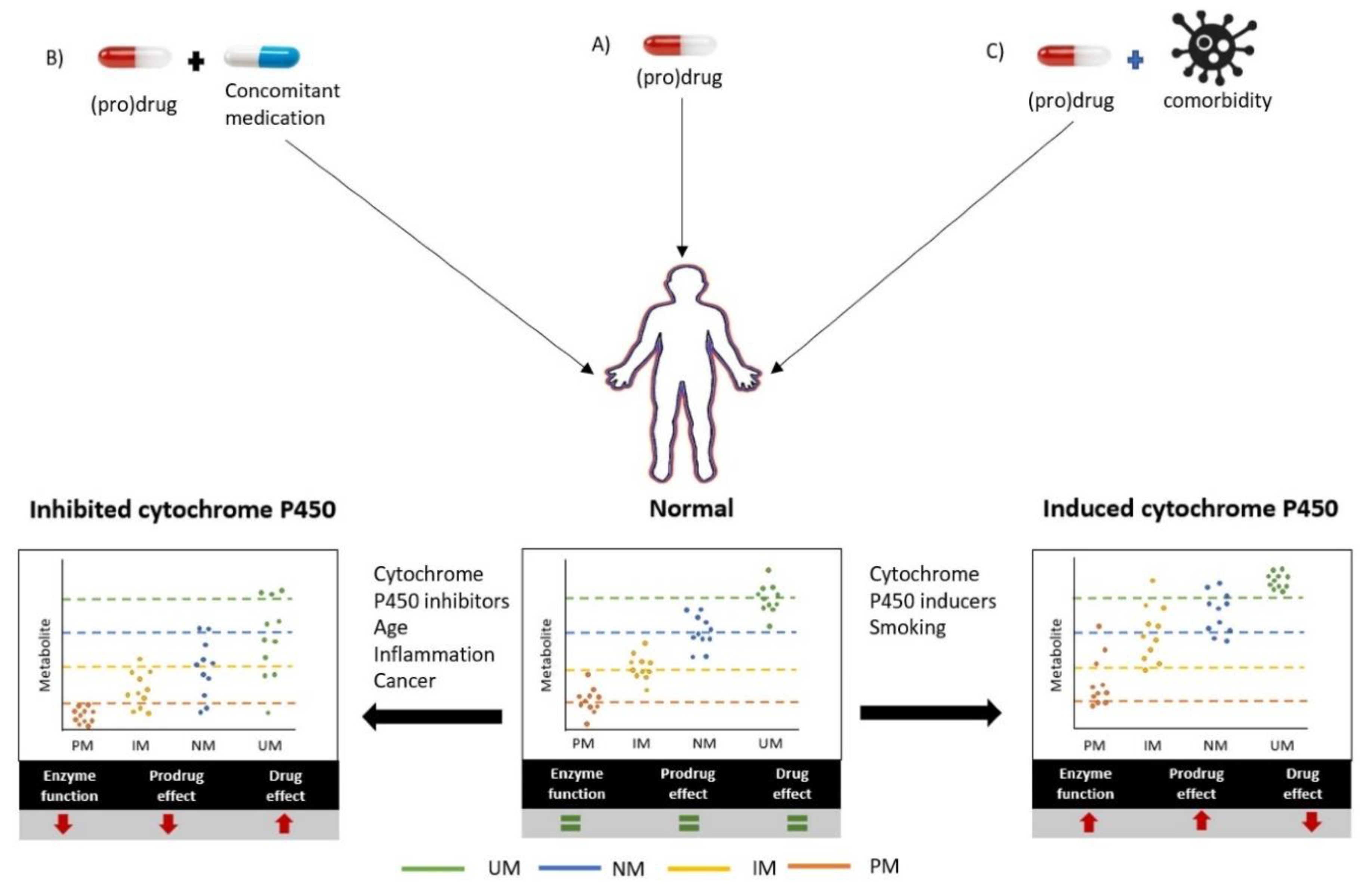
Figure 2.
Cytochrome P450-mediated drug metabolism can be inhibited or induced by the presence of co-medication and/or comorbidities, for which the magnitude of the changes depends on the host’s genotype. The metabolite concentrations in blood are plotted for the genotype-based predicted phenotypes (UM, NM, IM, and PM). This is presented for the normal situation (A) and for scenarios in which (B) CYP450 activity is inhibited by concomitant medication or (C) induced by a comorbidity. The resulting effects on drug efficacy are opposing for drugs and prodrugs. UM = ultra-rapid metabolizer, NM = normal metabolizer, IM = intermediate metabolizer, PM = poor metabolizer.
Figure 2.
Cytochrome P450-mediated drug metabolism can be inhibited or induced by the presence of co-medication and/or comorbidities, for which the magnitude of the changes depends on the host’s genotype. The metabolite concentrations in blood are plotted for the genotype-based predicted phenotypes (UM, NM, IM, and PM). This is presented for the normal situation (A) and for scenarios in which (B) CYP450 activity is inhibited by concomitant medication or (C) induced by a comorbidity. The resulting effects on drug efficacy are opposing for drugs and prodrugs. UM = ultra-rapid metabolizer, NM = normal metabolizer, IM = intermediate metabolizer, PM = poor metabolizer.


{kind=link}
{kind=link}
Table 1.
The impact of phenoconversion on CYP450 metabolism that results from the use of concomitant drugs and other extrinsic factors.
Table 1.
The impact of phenoconversion on CYP450 metabolism that results from the use of concomitant drugs and other extrinsic factors.
| Pharmacological Class | Drug | Enzyme | Cause of Phenoconversion | Type of Interaction | Drug/Factor Responsible for Phenoconversion | Type of Study | Genotype Assessment | Phenotype Assessment | Effect of Phenoconversion | Subjects | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Anticonvulsants | Valproate | CYP2C19 | Comedication | Inhibition | Stiripentol | 2 | CYP2C9*1/*2/*3 | Concentration-to-dose ratio of valproate | NM: Valproate serum concentration ↑↑ | 28 | [21] |
| CYP2C19*1/*2/*3 | PM: Valproate serum concentration ↑ | (Dravet syndrome patients) | |||||||||
| Clobazam | CYP2C19 | Comedication | Inhibition | Stiripentol/zonisamide | 2 | CYP2C19*1/*2/*3 | Clobazam and N-desmethyl-clobazam via HPLC | 22 NM zonisamide and stiripentol users (total 97 NM patients): N-desmethyl-clobazam concentration dose ratio ↑ | 238 | [22] | |
| NM: *1/*1 | 25 IM zonisamide and stiripentol users (total 133 IM patients): N-desmethyl-clobazam concentration dose ratio ↑ | (epilepsy patients) | |||||||||
| IM: *1/*2 or *1/*3 | 17 PM zonisamide and stiripentol users (total 72 PM patients): N-desmethyl-clobazam concentration dose ratio = | ||||||||||
| PM: *2/*2, *2/*3 or *3/*3 | |||||||||||
| Clobazam | CYP2C19 | Comedication | Induction | Phenytoin/carbamazepine | 2 | CYP2C19*1/*2/*3 | Clobazam and N-desmethyl-clobazam via HPLC | 51 NM phenytoin/carbamazepine users (total 97 NM patients): clobazam concentration dose ratio ↓ | 238 | [22] | |
| NM: *1/*1 | N-desmethyl-clobazam concentration dose ratio ↑ | (epilepsy patients) | |||||||||
| IM: *1/*2 or *1/*3 | 75 IM phenytoin/carbamazepine users (total 133 IM patients): clobazam concentration dose ratio ↓ | ||||||||||
| PM: *2/*2, *2/*3 or *3/*3 | N-desmethyl-clobazam concentration dose ratio ↑ | ||||||||||
| 36 PM phenytoin/carbamazepine users (total 72 PM patients): clobazam concentration dose ratio ↓ | |||||||||||
| N-desmethyl-clobazam concentration dose ratio = | |||||||||||
| Mephenytoin | CYP2C19 | Dietary | Inhibition | Alcohol consumption | 2 | CYP2C19*1/*2/*3/*4/*17 | Mephenytoin 4′-hydroxylation and mephenytoin via HPLC | The genotype-predicted phenotype overestimated (47%) the measured phenotype. For the overestimated group, 7 were alcohol users, resulting in lower CYP2C19 activity. | 114 | [8] | |
| PM: two loss of function alleles (*2, *3 or *4) | CYP2C19 activity: | (liver microsomes) | |||||||||
| IM: one functional and one loss of function allele | PM: <8 pmol/(mg protein*min) | ||||||||||
| NM: *1/*1 | UM: >75 pmol/(mg protein*min) | ||||||||||
| UM: one or two *17 alleles | IM: between 8 and 23 pmol/(mg protein*min) | ||||||||||
| (except *2/*17: IM or NM are accepted) | NM: between 23 and 75 pmol/(mg protein*min) | ||||||||||
| Anticoagulants | Warfarin | CYP2C19 | Comedication | Inhibition | Lansoprazole | 2 | CYP2C19 NM, IM and PM | * | There were no bleeding events in the rabeprazole control group (n = 41). Percentage of bleeding events in lansoprazole group (n = 41): | 82 | [23] |
| NM: 12% ↑ | (open heart surgery patients) | ||||||||||
| IM: 40% ↑↑ | |||||||||||
| PM: 22% ↑ | |||||||||||
| Showing de interaction between warfarin, lansoprazole and CYP2C19, (DDGI) | |||||||||||
| Antihypertensive | Debrisoquine | CYP2D6 | Comedication | Inhibition | Thioridazine | 1 | CYP2D6*1/*3/*4/*5/*6 | Phenotyping by debrisoquine, debrisoquine and 4-hydroxydebrisoquine ratios via GC | 8 NM→PM | 16 | [24] |
| 2 PM = PM | (psychiatric patients) | ||||||||||
| Antimuscarinics | Tolterodine | CYP2D6 | Comedication | Inhibition | Fluoxetine | 2 | CYP2D6*1/*3/*4 | Dealkyl and carboxyl tolterodine LC-ESI-MS/MS | NM: oral clearance of tolterodine ↓ (80%) | 9 | [25] |
| Fluoxetine and norfluoxetine via HPLC-UV | IM: oral clearance of tolterodine ↓↓ (93%) | (depressed patients) | |||||||||
| Possible: NM→PM | |||||||||||
| Antipsychotics | Aripiprazole, haloperidol, risperidone, paliperidone zuclopenthixol | CYP2D6 | Comedication | Inhibition | Strong inhibitors (paroxetine/bupropion) | 1 | CYP2D6*1/*2/*3/*4/*5/*6/*7/*8/*9/*10/*11/*15/*17/*29/*41, deletions and duplications | Concentration of parent compounds antipsychotics via HPLC-MS/MS | 8 patients taking CYP2D6 inhibitors | 82 | [26] |
| Moderate inhibitors (sertraline/duloxetine) | Strong inhibitors: | (psychiatric patients) | |||||||||
| 4 NM→PM | |||||||||||
| 1 IM→PM | |||||||||||
| Moderate inhibitors: | |||||||||||
| 3 NM→IM | |||||||||||
| Aripiprazole | CYP2D6 | Comedication | Inhibition | CYP2D6 inhibitors | 2 | CYP2D6*1/*3/*4/*5/*6/*10/*41 and duplication, CYP3A4*1/*1B/*22, and CYP3A5*1/*3 | Aripiprazole, dehydroaripiprazole, N-dealkyl-aripiprazole, monohydroxy-aripiprazole and dehydroaripiprazole via LC-MS/MS | NM: aripiprazole concentration ↑↑ (50%) | 93 | [27] | |
| UM: aripiprazole concentration ↑ (20%) | (psychiatric patients) | ||||||||||
| Olanzapine | CYP1A2 | Smoking | Induction | Polycyclic aromatic hydrocarbons | 2 | CYP2D6*1/*3/*4/*5/*6/*41 and duplication | Olanzapine and desmethylolanzapine via LD-MS | CYP1A2*1F/*1F smokers: 4′-N-desmethylolanzapine/olanzapine ratio ↑: | 37 | [28] | |
| PM: two defective alleles (*3, *4, *5 or *6 ) | 132% compared to smokers with other CYP1A2 polymorphisms and, 107% compared to nonsmokers that are homozygous carriers of CYP1A2*1F | (schizophrenia or schizoaffective disorder patients) | |||||||||
| IM: one defective allele | |||||||||||
| NM: *1/*1 | |||||||||||
| UM: gene duplication together with *1 | |||||||||||
| ABCB1 | |||||||||||
| CYP1A2*1/*1C/*1D/*1K/*1F | |||||||||||
| Antitussive and pain medication | Dextromethorphan, tramadol | CYP2D6 | Comedication | Inhibition | Duloxetine/paroxetine | 1 | CYP2D6*1/*2/*3/*4/*6/*7/*8/*9/*10/*17/*29/*35/*41/*5 (deletion) and duplication | Phenotyping of dextromethorphan and tramadol by LC–MS/MS followed by LLE | Duloxetine: | 17 | [29] |
| PM: UMRDEM/DOR > 0.3 | 71% NM→IM | (healthy volunteers) | |||||||||
| IM: 0.03 < UMR < 0.3 | 25% NM→PM | ||||||||||
| NM: 0.003 < UMR < 0.03 | Paroxetine: | ||||||||||
| UM: UMR < 0.003 | 94% heterozygous NM→PM | ||||||||||
| 56% homozygous NM→PM | |||||||||||
| Dextromethorphan | CYP2D6 | Comedication | Inhibition | Levomeprazine/biperiden | 1 | CYP2D6*1/*2/*3/*4/*5/*8/*10/*18/*21 | Dextromethorphan and dextrorphan via HPLC | 14 patients: | 104 | [6] | |
| Levomepromazine and biperiden via GCMS | 3 NM→PM | (90 healthy volunteers) | |||||||||
| 2 NM→IM | (14 schizophrenia patients) | ||||||||||
| 9 stayed IM | |||||||||||
| Cardiac drugs | Flecainide | CYP2D6 | Comedication | Inhibition | Bepridil | 1 | CYP2D6*1/*2/*4/*5/*10/*14/*21/*36 | Flecainide enantiomers via HPLC | All 17 concomitant treated NM patients showed a serum flecainide S/R ratio of PM subjects: NM→PM | 143 | [22] |
| NM: *1 and *2 | (supraventricular tachyarrhythmia patients) | ||||||||||
| IM: *10 | |||||||||||
| PM: *4/*5/*14/*21/*36 | |||||||||||
| NSAID | Flurbiprofen | CYP2C9 | Comedication | Inhibition | Fluconazole | 1 | CYP2C9*1/*2/*3 | Flurbiprofen, 4′-hydroxyflurbiprofen and fluconazole via HPLC | 200 mg fluconazole: | 189 | [30] |
| 11 NM→IM | (healthy subjects) | ||||||||||
| 8 IM→PM | |||||||||||
| 2 PM = PM | |||||||||||
| 400 mg fluconazole: | |||||||||||
| 11 NM→PM | |||||||||||
| 8 IM→PM | |||||||||||
| 2 PM = PM | |||||||||||
| Proton pump inhibitors | Lansoprazole | CYP2C19 | Comedication | Inhibition | Fluvoxamine | 2 | CYP2C19*1/*2/*3 | Lansoprazole enantiomers and lansoprazole sulphone via HPLC | NM: (R)-lansoprazole AUC ↑↑ (903%) | 18 | [31] |
| NM: *1/*1 | (S)-lansoprazole AUC ↑↑ (1664%) | (healthy volunteers) | |||||||||
| IM: *1/*2 or *1/*3 | IM: (R)-lansoprazole AUC ↑ (462%) | ||||||||||
| PM: *2/*2 or *2/*3 | (S)-lansoprazole AUC ↑ (781%) | ||||||||||
| Antiestrogens | Tamoxifen | CYP2D6 | Comedication | Inhibition | Potent inhibitors: paroxetine and fluoxetine | 1 | CYP2D6*1/*3/*4/*6/*7/*8/*10/*11/*14/*15/*17/*19/*20/*25/*26/*29/*31/*35/*36/*40/*41 and duplications | Tamoxifen and metabolites via HPLC | 42 CYP2D6 subjects with CYP2D6 inhibitor compared to 85 subjects without concomitant medication. | 158 | [32] |
| Weak inhibitors: sertraline, citalopram, celecoxib, diphenhydramine, and chlorpheniramine | Mean group endoxifen plasma concentrations by potent inhibitor usage: | (breast cancer patients) | |||||||||
| 1 UM→IM | |||||||||||
| 5 NM→PM | |||||||||||
| 11 IM→PM | |||||||||||
| Mean group endoxifen plasma concentrations by weak inhibitor usage: | |||||||||||
| 3 UM→NM or IM | |||||||||||
| 10 NM→IM | |||||||||||
| 12 IM→PM | |||||||||||
| Tamoxifen | CYP3A4 | Other | Induction | Vitamin D | 2 | CYP3A4*1/*22, POR*28, CYP2C9*1/*2/*3, CYP2B6*1/*4/*5/*6, MDR1 c.3435C > T, BCRP c.421C > A/c.34G > A, CYP3A5*1/*3, CYP2D6*1/*3/*4/*5/*9/*10/*41 | Tamoxifen, NDM-tamoxifen, 4-OH-tamoxifen, Z-endoxifen, Z-3-OH tamoxifen and Z-α-OH-tamoxifen via LC-MS/MS normalized to 4-β-OH-cholesterol via UHPLC-MS/MS and 25-OH-vitamin D via ELISA | During winter months endoxifen levels ↓ (20%) | 196 | [33] | |
| (breast cancer patients) |
Type of study refers to “designed with the objective to study phenoconversion” (Type 1) or “not specifically designed to study phenoconversion, did not categorize patients into PM, IM, NM, RM or UM phenotype” (Type 2). AUC = area under the curve, LC–ESI–MS/MS = liquid chromatography electrospray ionization tandem mass spectrometric, HPLC–UV = high pressure liquid chromatography coupled ultraviolet, HPLC–MS/MS = high pressure liquid chromatography tandem mass spectrometry, LLE = liquid–liquid extraction, GCMS = gas chromatography mass spectrometry, LD–MS = laser-assisted desorption mass spectrometry, UHPLC–MS/MS = = ultrahigh pressure liquid chromatography tandem mass spectrometry, ELISA = enzyme-linked immuno sorbent assay, UMR = urinary metabolic ratio, UM = ultra-rapid metabolizer, NM = normal metabolizer, IM = intermediate metabolizer, PM = poor metabolizer, “→” = converted to, “↑” = increase, “↑↑” = larger increase, “↓” = decrease, “↓↓” = larger decrease, “=” = did not change. *This study did not perform phenotype assessment; nevertheless, the study showed the impact of phenoconversion and, therefore, the study was included in this systemic review. The impact of phenoconversion is analyzed via hemorrhagic complications (minor bleeding events such as tarry stool, bleeding from colon diverticulum, conjunctival bleeding, or bleeding into the shoulder joint or surgical sites).
Table 2.
The impact of phenoconversion on CYP450 metabolism that results from the use of patient- and disease-related factors.
Table 2.
The impact of phenoconversion on CYP450 metabolism that results from the use of patient- and disease-related factors.
| Cause of Phenoconversion | Drug | Enzyme | Result | Type of Study | Genotype Assessment | Phenotype Assessment | Effect of Phenoconversion | Subjects | Ref |
|---|---|---|---|---|---|---|---|---|---|
| Age | Omeprazole | CYP2C19 | Reduced activity | 1 | CYP2C19*1/*2/*3 | Omeprazole (10 mg for elderly and 20 mg for young), omeprazole, 5-hydroxyomeprazole and omeprazole sulfone via LC | 38% NM→PM | 51 | [34] |
| 42% IM→PM | (healthy volunteers: older age | ||||||||
| (elderly 66–85 years, younger 21–36)) | |||||||||
| Cancer | Omeprazole | CYP2C19 | Reduced activity | 1 | CYP2C19*1/*2 | Phenotyping via the prodrug omeprazole (20 mg) measured in plasma. | 25% NM→PM | 16 | [35] |
| NM: log10 [OM]/[OH-OM] <1 | (advanced cancer patients) | ||||||||
| PM: log10 [OM]/[OH-OM] ≥1 | |||||||||
| Omeprazole | CYP2C19 | Reduced activity | 1 | CYP2C19*1/*2/*3 | Phenotyping via the prodrug omeprazole (20 mg) measured in plasma. | 37% NM→PM | 33 | [36] | |
| NM: log10 [OM]/[OH-OM] <1 | (30 NM patients) | (advanced cancer patients) | |||||||
| PM: log10 [OM]/[OH-OM] ≥1 | |||||||||
| Proguanil | CYP2C19 | Reduced activity | 1 | CYP2C19*1/*2/*3/*17 | Proguanil (PG) (200 mg), PG and cycloguanil (CG) via HPLC | 27% NM→PM | 25 | [37] | |
| NM: PG/CG ≥ 1 | 53% IM→PM | (multiple myeloma patients) | |||||||
| PM: PG/CG < 1 | |||||||||
| Inflammation | Losartan | CYP2C9 | Reduced activity | 1 | CYP2C9*1/*2/*3 | Losartan and metabolite (E-3174) via HPLC | Mean group metabolic ratio in patients: | 51 | [38] |
| 31 NM→IM | (patients with Behçet’s disease) | ||||||||
| 20 IM→PM | 96 | ||||||||
| (healthy volunteers) | |||||||||
| Omeprazole | CYP2C19 | Reduced activity | 1 | CYP2C19*1/*2/*3 | Omeprazole and 5--hydroxy omeprazole via LC | Mean group metabolic ratio of omeprazole/5-hydroxy omeprazole in patients: | 31 | [39] | |
| NM: *1/*1 | NM→PM | (patients with chronic hepatitis or cirrhosis that tested positive for hepatitis C virus (HCV)) | |||||||
| IM: *1/*2 or *1/*3 | IM→PM | 30 | |||||||
| PM: *2/*2, *2/*3 or *3/*3 | PM = PM | (healthy volunteers) | |||||||
| Voriconazole | CYP2C19 | Reduced activity | 2 | CYP2C19*1/*2/*3/*17 | Voriconazole and voriconazole-N-oxide concentrations were measured via LC-MS/MS | 20 patients applicable for genotyping: | 36 | [40] | |
| UM: *1/*17 | Metabolic ratio: | (patients treated with intravenous or oral voriconazole) | |||||||
| NM: *1/*1 | UM = −0.994147N | ||||||||
| IM: *1/*2 or *1/*3 | NM = −0.991972N | ||||||||
| PM: *2/*2, *2/*3 or *3/*3 | IM = −0.986512N | ||||||||
| Voriconazole trough concentration: | |||||||||
| UM = +1.003685N | |||||||||
| NM = +1.004965N | |||||||||
| IM = +1.009365N | |||||||||
| N = CRP concentration | |||||||||
| Mephenytoin | CYP2C19 | Reduced activity | 2 | CYP2C19*1/*2/*3/*4/*17 | Mephenytoin 4′-hydroxylation and mephenytoin via HPLC | The genotype-predicted phenotype overestimated (47%) the measured phenotype. For the overestimated group, 9 had inflammatory diseases (rheumatoid arthritis and gastrointestinal perforation), resulting in lower CYP2C19 activity. | 114 | [8] | |
| PM: two loss of function alleles (*2, *3 or *4) | CYP2C19 activity: | (liver microsomes) | |||||||
| IM: one functional and one loss of function allele | PM: <8 pmol/(mg protein*min) | ||||||||
| NM: *1/*1 | UM: >75 pmol/(mg protein*min) | ||||||||
| UM: one or two *17 alleles | IM: between 8 and 23 pmol/(mg protein*min) | ||||||||
| (except *2/*17: IM or NM is accepted) | NM: between 23 and 75 pmol/(mg protein*min) | ||||||||
| Dextromethorphan | CYP2D6 | Reduced activity- | 1 | CYP2D6 | Dextromethorphan (DEM) (2.5 mg), DEM and dextrorphan (DOR) via HPLC | 10 LKM-1 positive subjects: | 1723 | [41] | |
| 33 allelic variants (AmpliChip®) | UM: DEM/DOR < 0.003 | 6 NM→IM | (chronic hepatitis C patients) | ||||||
| NM: 0.003 < DEM/DOR < 0.03 | 1 NM→PM | ||||||||
| IM: 0.03 < DEM/DOR < 0.3 | CYP2D6 metabolic function is reduced by LK-1 antibody presence | ||||||||
| PM: DEM/DOR > 0.3 | |||||||||
| Immunosuppressants (tacrolimus or cyclosporine A) | CYP3A5 | Reduced activity | 1 | CYP3A5*1/*3 | 4β-hydroxycholesterol via GCMS | 10 CYP3A5*1 recipients had low CYP3A5 activity (phenoconversion), might be due to increased indoxyl sulfate concentrations | 63 | [42] | |
| (kidney transplant recipients) | |||||||||
| Pregnancy | Dextromethorphan | CYP2D6 | Reduced activity | 2 | CYP2D6*1/*3/*4 | Dextromethorphan (30 mg) | NM: metabolic ratio dextromethorphan/dextrorphan ↓ (29%) | 140 | [43] |
| CYP2D6: O-demethylated → dextrorphan (dextromethorphan/dextrorphan) | IM: metabolic ratio dextromethorphan/dextrorphan ↓ (63%) | (pregnant women) | |||||||
| CYP3A: N-demethylated to 3-methoxymorphinan (dextromethorphan/3-demethoxymorphinon) | |||||||||
| Paroxetine | CYP2D6 | Increased activity | 2 | CYP2D6*1/*3/*4/*5/*6/*9/*10/*41 and duplication | Paroxetine via HPLC-UV or LC-MS/MS | NM/UM: plasma paroxetine concentration ↓ | 74 | [44] | |
| NM: *1/*1 | PM plasma paroxetine concentration ↑ | (pregnant women) | |||||||
| UM: duplication | |||||||||
| IM: heterozygous for nonfunctional allele | |||||||||
| PM: homozygous for nonfunctional allele |
Type of study refers to “designed with the objective to study phenoconversion” (Type 1) or “not specifically designed to study phenoconversion, did not categorize patients into PM, IM, NM, RM or UM phenotype” (Type 2). HPLC–MS/MS = high pressure liquid chromatography tandem mass spectrometry, GCMS = gas chromatography mass spectrometry, ELISA = enzyme-linked immuno sorbent assay, UM = ultra-rapid metabolizer, NM = normal metabolizer, IM = intermediate metabolizer, PM = poor metabolizer, “→” = converted to, “↑” = increase, “↑↑” = larger increase, “↓” = decrease, “↓↓” = larger decrease.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Klomp, S.D.; Manson, M.L.; Guchelaar, H.-J.; Swen, J.J. Phenoconversion of Cytochrome P450 Metabolism: A Systematic Review. J. Clin. Med. 2020, 9, 2890. https://doi.org/10.3390/jcm9092890
AMA Style
Klomp SD, Manson ML, Guchelaar H-J, Swen JJ. Phenoconversion of Cytochrome P450 Metabolism: A Systematic Review. Journal of Clinical Medicine. 2020; 9(9):2890. https://doi.org/10.3390/jcm9092890
Chicago/Turabian StyleKlomp, Sylvia D., Martijn L. Manson, Henk-Jan Guchelaar, and Jesse J. Swen. 2020. "Phenoconversion of Cytochrome P450 Metabolism: A Systematic Review" Journal of Clinical Medicine 9, no. 9: 2890. https://doi.org/10.3390/jcm9092890
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.