Article Text

Abstract
Background PTEN hamartoma tumour syndrome (PHTS) encompasses several clinical syndromes with germline mutations in the PTEN tumour suppressor gene, including Cowden syndrome which is characterised by an increased risk of breast and thyroid cancers. Because PHTS is rare, data regarding cancer risks and genotype–phenotype correlations are limited. The objective of this study was to better define cancer risks in this syndrome with respect to the type and location of PTEN mutations.
Methods 154 PHTS individuals with a deleterious germline PTEN mutation were recruited from the activity of the Institut Bergonié genetic laboratory. Detailed phenotypic information was obtained for 146 of them. Age and sex adjusted standardised incidence ratio (SIR) calculations, cumulative cancer risk estimations, and genotype–phenotype analyses were performed.
Results Elevated SIRs were found mainly for female breast cancer (39.1, 95% CI 24.8 to 58.6), thyroid cancer in women (43.2, 95% CI 19.7 to 82.1) and in men (199.5, 95% CI 106.39 to 342.03), melanoma in women (28.3, 95% CI 7.6 to 35.4) and in men (39.4, 95% CI 10.6 to 100.9), and endometrial cancer (48.7, 95% CI 9.8 to 142.3). Cumulative cancer risks at age 70 were 85% (95% CI 70% to 95%) for any cancer, 77% (95% CI 59% to 91%) for female breast cancer, and 38% (95% CI 25% to 56%) for thyroid cancer. The risk of cancer was two times greater in women with PHTS than in men with PHTS (p<0.05).
Conclusions This study shows a considerably high cumulative risk of cancer for patients with PHTS, mainly in women without clear genotype–phenotype correlation for this cancer risk. New recommendations for the management of PHTS patients are proposed.
- Cancer: Breast
- Clinical Genetics
- Guidelines
- Molecular Genetics
- Thyroid Disease
Statistics from Altmetric.com
Introduction
Cowden syndrome (CS) is an autosomal dominant inherited disorder characterised by macrocephaly, benign lesions that can develop in many organs, and an increased risk of breast, thyroid and possibly other cancers.1 It is allelic to other seemingly unrelated clinical syndromes including the paediatric form Bannayan–Riley–Ruvalcaba syndrome (BRRS), Lhermitte–Duclos disease (LDD), SOLAMEN (segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus) syndrome, macrocephaly/autism syndrome, and juvenile polyposis syndrome,2–6 which are collectively referred to as PTEN hamartoma tumour syndrome (PHTS).7
The PTEN gene, located on chromosome 10q23.3, encodes a dual specificity phosphatase that plays a tumour suppression role by negatively regulating the PI3K/Akt/mTOR cell survival signalling pathway.8 The PTEN structure reveals two main domains: the phosphatase domain (aa 7–185) which contains the PTEN active site in exon 5; and the C2 domain (aa 186–351) which binds phospholipids and positions the catalytic domain on the membrane.9
Although it is generally accepted that CS patients show an elevated risk for breast and thyroid cancers, with risks generally being reported around 25–50% for breast cancer and 3–10% for thyroid cancer,10–12 more recent studies have revealed a higher risk for breast and thyroid cancers and have extended this elevated risk to other cancer sites including endometrial, kidney, colorectal cancers, and melanoma.13 ,14
Because CS is rare and diagnosis difficult due to highly variable expressivity, data regarding genotype–phenotype correlations are limited and mostly based on a collection of case series, each generally comprising a small number of patients. To date, most studies have failed to demonstrate a consistent genotype–phenotype correlation in CS.15–18 This study aims to clarify these issues by: (1) collecting phenotypic and molecular data in a large series of PHTS patients sharing a deleterious germline PTEN mutation; (2) assessing cancer risks in these patients; and (3) investigating if a correlation exists between genotype and cancers, but also between genotype and non-malignant findings of the PHTS. The information generated by our data will also allow us to clarify and adapt surveillance recommendations.
Patients and methods
Patients
Between January 1997 and December 2008, 546 patients were referred for PTEN mutation analysis to the Institut Bergonié genetic laboratory (Bordeaux, France) as part of a molecular diagnosis of PHTS. Of these 546 patients, 99 were shown to be carriers of a deleterious germline PTEN mutation and were selected for the study. Patients with a typical CS phenotype without identified PTEN mutations were excluded. Among families of selected patients, 55 supplementary carriers of a deleterious germline PTEN mutation were identified by genetic testing. In all cases, the PTEN mutation search was made after obtaining a signed informed consent in the context of a medical genetic diagnosis of suspected PHTS, in compliance with the French law on genetic testing (law number 94–654).
From the 154 selected patients, detailed phenotypic information was obtained for 146 who constitute the main study population. These patients were from 98 distinct PHTS families. Most of them lived in France (96% (132/146)); the others were Spanish, Portuguese, Swiss, Algerian and Tunisian. There were 70 women and 76 men, with a median age of 36 years (range 6 months–76 years), and 27% (39/146) of these patients were under 20 years old.
Finally, from selected patients, three kinds of indications for PTEN analysis could be distinguished: (1) various findings evocative of PHTS without associated cancer (n=63, 43%); (2) various findings evocative of PHTS with one or more cancers (n=33, 23%); and (3) genetic tests in family members in which a PTEN mutation had been previously identified (n=50, 34%). A PTEN analysis was never requested when there was only the presence of cancer in the index case and in his/her family as no mutations have been found with this indication.
Molecular analysis
PTEN mutation analysis was performed with a combination of denaturing gradient gel electrophoresis, denaturing high performance liquid chromatography, and direct Sanger sequencing to detect point mutations, and with a combination of multiplex amplifiable probe hybridisation and quantitative multiplex PCR of short fluorescent fragments to detect large deletions.
Clinical information questionnaires
To obtain clinical background information on these patients, we sent a clinical information questionnaire to the various physicians referring patients for PTEN mutation analysis (eg, geneticists, paediatricians, and dermatologists). The questionnaire was composed of 10 parts that investigated the manifestations of CS and the date of their occurrence: age of diagnosis, last news and death; mucocutaneous lesions; gastrointestinal lesions; thyroid lesions; breast lesions; macrocephaly (occipitofrontal circumference >97th centile); brain lesions, lipomatosis and vascular lesions, genitourinary lesions; other cancers.
Statistical analysis
Standardised incidence ratios (SIR) were calculated by an indirect method19 using age and sex adjusted specific incidence data from the French cancer registries (year 2005).20 There were 28 categories on the basis of two genders and 14 age groups. Incidence was assumed to be 0 for categories where statistics were not provided. Person-years of observation (PYO) were calculated for each type of cancer from birth date to the date of cancer for individuals who developed cancer, or to the date of the most recent information for individuals without cancer. The expected number of cancers was calculated by multiplying cancer incidence rates in each of the 28 categories by PYO in each category (indirect standardisation). These analyses were performed using a 95% CI and χ2 tests.
Risks of cancer by age were estimated using Kaplan–Meier survival analysis. Follow-up time for every patient was calculated as the time between the date of birth and the date of cancer diagnosis, last contact, or death. It should be noted that the date of cancer diagnosis was missing for two patients, so in order not to exclude them from the cancer risk calculations, the age at last news was taken as a substitute. We used the log rank test for comparisons of cancer risks according to the sex of the patients and according to genotype. All analyses were carried out with Stata software (Statacorp, College Station, Texas, USA).
χ2 or Fisher tests were performed to analyse correlations between phenotypes and genotypes. Genotypes were classified according to mutation types (truncated vs non truncated mutations) and to the protein domains (phosphatase vs C2 domain). It should be noted that for the phenotype study, according to the protein domain implicated, we had to exclude six patients for whom the presence of a large PTEN deletion affected the two domains studied and ruled out any correlation analyses. Phenotypes were defined according to the questionnaire responses. Individuals under 20 years of age were excluded from the analyses of mucocutaneous lesions (n=39). This meant that the calculation was not affected by age at diagnosis and enabled us to carry out correlation analyses between the genotype and the different cutaneous lesions. Concerning the genital pigmentation analysis, it was performed in all men without reference to age because, if it existed, it was present in the early years of life.
A value of p<0.05 was considered significant for all statistical tests.
Results
Genotype
All patients had a pathogenic germline PTEN mutation; that is, 74 different mutations across the 146 patients (figure 1 and see online supplementary table S1). We observed 49 (34%) frameshift mutations, 44 (30%) missense mutations, 31 (21%) nonsense mutations, 15 (10%) splice mutations, and 7 (5%) large deletions. In total, 102 (70%) mutations were considered as truncated mutations in contrast to missense mutations.

Distribution of mutations in the PTEN gene. The spectrum of PTEN mutations reported was frameshift (blue), missense (red), nonsense (green), splice site (grey), and large deletion (black dashed line). The height of the full line is proportional to the mutation recurrence. ATG and TGA are, respectively, the start and stop codons in the PTEN genomic sequence.
The mutation spectrum for the 146 probands revealed mutations that were distributed irregularly across exons, with, in decreasing order, 46 (32%) mutations in exon 5, 28 (19%) in exon 8, 18 (12%) in exon 6, and 15 (10%) in exon 7. Few mutations were observed in exons 1 to 4 (between 2–4% according to the exons), but none in exon 9 (figure 1). Germline variations in the 5′ UTR of PTEN were excluded due to the uncertainties existing regarding their pathogenic character.
A total of 82 (59%) mutations occurred within the phosphatase domain, and among them, 12 (15%) concerned the amino acid 130 which is involved into the catalytic site (aa 123–130) of the PTEN protein. The others amino acids of the catalytic site were not concerned. A total of 58 (41%) mutations occurred within the C2 domain of the protein.
Phenotype
The clinical phenotype of each patient was analysed, criterion by criterion, to assess their respective prevalence (table 1). Missing data on questionnaire items are detailed in online supplementary table S1 but are not taken into account in the following calculations. The number of patients with information available is described in brackets for each item.
Genotype–phenotype analysis in 146 patients with PTEN hamartoma tumour syndrome
Overall, dermatologic features (98%), macrocephaly (93%), gastrointestinal polyps (93%), and breast (74%) and thyroid (71%) benign lesions were the most frequently observed features in our series.
Specifically, mucocutaneous pathognomonic criteria such as facial papules, oral papillomatosis and acral keratosis were found in more than 70% of patients. In contrast, multiple facial trichilemmomas were found in 38% (23/61) of patients and pigmented penile macules were noted in 48% (23/50) of men.
Gastrointestinal polyps were observed throughout the digestive tract. Colonic polyps were the most frequent, affecting 85% (67/79) of patients, with various histologies including: hamartomas (44%), lymphoid aggregates (35%), adenomas (34%), ganglioneuromas (25%), and lipomas (6%). Gastric polyps and glycogenic acanthosis of the oesophagus were respectively found in 73% (44/60) and 48% (28/58) of patients.
For breast lesions, fibrocystic disease was the most frequently reported, affecting 47% (26/55) of women in the study. Other benign lesions were less reported including papillomas, adenomas, hamartomas, lipomas, and a few cases of virginal breast hypertrophy. For the thyroid lesions, multiheteronodular goitres and adenomas were the most often reported, affecting respectively 62% (81/131) and 23% (30/131) of patients with a median age of 26 (range 6–66) years.
We also noted less prevalent clinical features such as lipomas (48%), vascular malformations (35%), and genitourinary lesions that were especially found in women (52%) with ovarian cysts (37%) and uterine fibromas (25%). For neurologic disorders, LDD was observed in 13% (19/144) of patients, and mental retardation—with or without autism—in 10% (15/144) of cases.
Furthermore, we noted the occurrence of 13 (9%) deaths during the study with a median age of 50 (range 28–71) years. Nine patients died after developing cancer, two of whom were young adults (28 and 33 years). Two other patients died of cardiac decompensation resulting from important high flow vascular malformations at age 29 and 32 years. The last two patients died of other causes at an older age.
Cancer description
Forty per cent (59/146) of patients developed cancer with a median age of 36 (range 4–63) years; 75% (44/59) of them had only one cancer, 20% (12/59) had two separate cancers, and 5% (3/59) had three separate cancers (table 1 and see online supplementary table S2).
The most commonly reported cancer for women was breast cancer (34%, n=23/68) with a median age of 42 (range 27–50) years. Of female CS patients diagnosed with breast cancer, 48% (11/23) had bilateral disease. In the 21 cases of breast cancer for whom histopathology analysis data were available, eight (38%) were intraductal carcinomas and 13 (62%) were invasive ductal carcinomas. Of these, six were associated with intraductal carcinomas and two were apocrine carcinomas.
The second most frequently reported cancer was thyroid cancer (17%, n=24/140) with a median age of 31 (range 16–57) years. Note that 10 of the 24 reported cases were precancerous lesions, including papillary microcarcinomas and minimally invasive follicular carcinomas. In the 22 cases of thyroid cancer with histopathology data available, 13 (59%) were papillary carcinomas, six (27.5%) were follicular carcinomas, one (4.5%) was a hybrid tumour between follicular and papillary types, one (4.5%) was a medullary carcinoma, and one (4.5%) was an oxyphil cell tumour. This distribution confirms the known relative overrepresentation of follicular carcinomas in CS, particularly for patients over 18 years of age,21 with six cases among the 20 patients over 18.
Few cases of cancer were reported for the other organs at risk. We observed 6% of melanoma (n=9), 3% of colorectal cancer (n=4), 3% of ovary cancer (n=4), 2% of endometrial cancer (n=3), and 2% of renal cell carcinoma (n=3). Note that three of the four ovary cancers were embryonic tumours. Other types of cancer were reported more sporadically (n≤2) such as testicular, lung, ethmoid, vocal cord, and parotid cancers, and basal cell carcinoma.
Cancer risks
An elevated risk of cancer was found overall for both women (age and sex adjusted SIR 22.9, 95% CI 16 to 31.6) and men (SIR 11.9, 95% CI 7.5 to 17.9) and especially for: female breast cancer (SIR 39.1, 95% CI 24.8 to 58.6); thyroid cancer in women (SIR 43.2, 95% CI 19.7 to 82.1) and in men (SIR 199.5, 95% CI 106.4 to 342); endometrial cancers (SIR 48.7, 95% CI 9.8 to 142.3); melanoma in women (SIR 28.3, 95% CI 7.6 to 35.4) and in men (SIR 39.4, 95% CI 10.6 to 100.9); and for kidney cancer in women (SIR 48.9, 95% CI 5.5 to 176.5). Age and sex adjusted SIRs were not statistically significant for colorectal cancer for women or men, nor was kidney cancer for men (table 2).
Standardised incidence rates (SIR) of cancer in the patient population
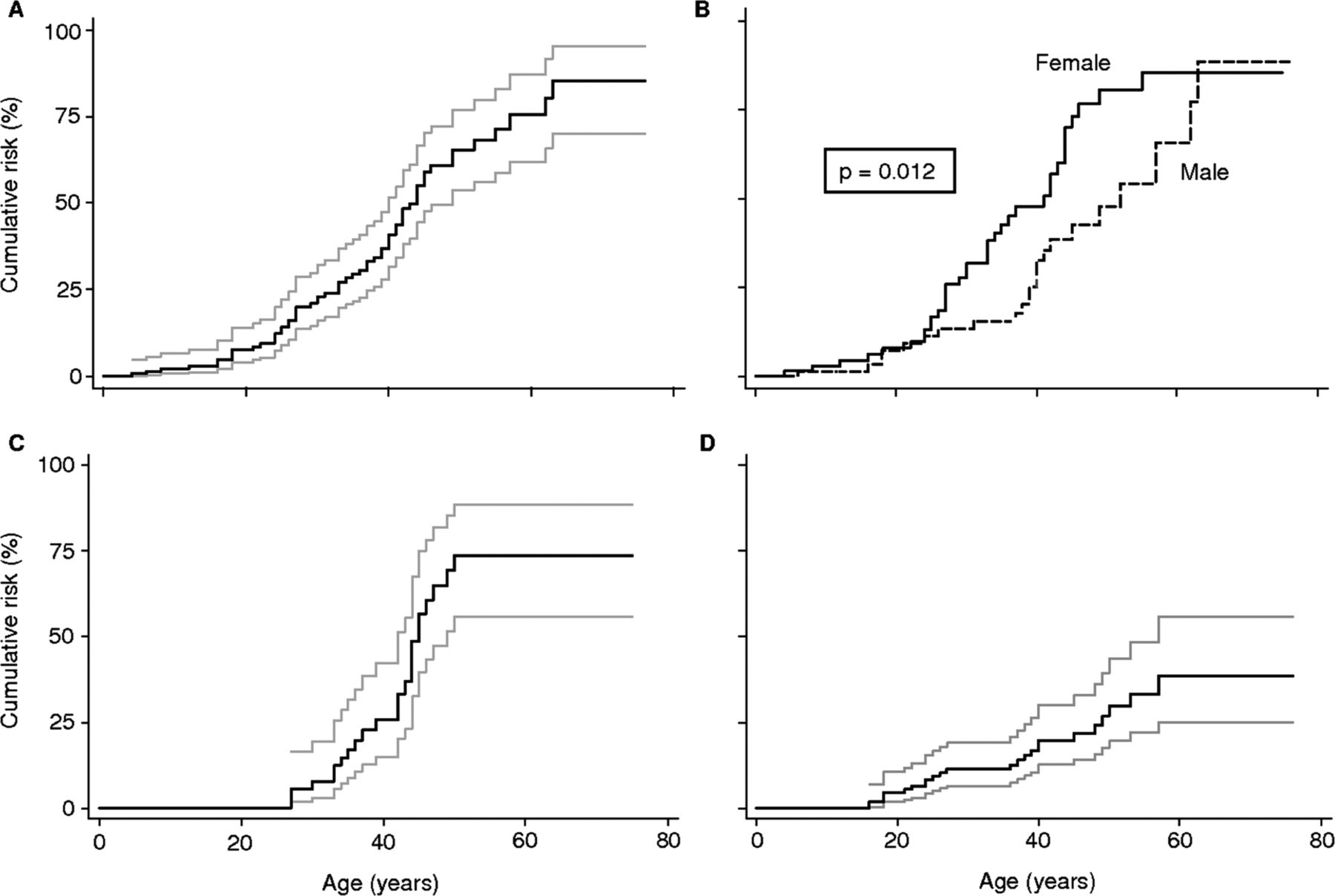
We observed that the cumulative risk of cancer at age 70 (for any cancer) was 85% (95% CI 70% to 95%), and that this risk was significantly higher (p=0.01) among women at age 50 (81%, 95% CI 65% to 92%) than among men at age 50 (48%, 95% CI 32% to 66%). This confirms the results obtained by the calculation of the SIR above, showing that the risk of developing any cancer was two times greater in women (SIR 22.9) than in men (SIR 11.9). The cumulative cancer risk at age 70 was 77% (95% CI 59% to 91%) for female breast cancer and 38% (95% CI 25% to 56%) for thyroid cancer across both genders (table 3 and figure 2). Note that the evaluation of the cancer risk by the Kaplan–Meier method was carried out only for cancer in general, and breast and thyroid cancers, due to the low number of cases reported for the other types of cancer (see online supplementary table S2).
Cumulative cancer risk* by age and type of cancer in 146 patients with PTEN hamartoma tumour syndrome

{kind=link}
{kind=link}
Kaplan–Meier estimation of cumulative lifetime cancer risks in patients with PTEN hamartoma tumour syndrome or Cowden syndrome (grey lines: 95% CIs). (A) Cumulative risk of any cancer. (B) Cumulative risk of any cancer according to sex (full line: female; dashed line: male; p: log rank test). (C) Cumulative risk of female breast cancer. (D) Cumulative risk of thyroid cancer.
Genotype–phenotype correlations
No significant correlation (p>0.05) was found between the occurrence of the different types of cancer that were investigated and the mutation type (truncated mutations vs no truncated mutations) or the protein domain (phosphatase domain vs C2 domain). These results are reported in online supplementary figures (S1–S6).
No significant correlation (p>0.05) has been demonstrated between the non-malignant clinical features of PHTS that were investigated and the mutation type or the protein domain. These results are reported in table 1.
Discussion
We report the phenotypic evaluation of 146 patients carrying a deleterious germline PTEN mutation and we observe a considerably high cumulative risk of cancer, especially in women. No genotype–phenotype correlations for malignant pathologies or for findings of the disease were observed.
Cancer risks
This study underlines the severity of cancers in CS and provides estimations of the cancer risk. Overall, 40% of patients in this study developed a cancer, with one quarter developing tumours across two or three different sites. Cancer was the main cause of death during this study with a young median age at death of 36 years.
In accordance with other reports,1 the two most frequently reported cancers were female breast cancer, with bilateral tumours in almost half of the cases, and thyroid cancer. For both of these sites, median age at diagnosis was young, with the youngest patients in our series being 27 and 16 years old, respectively.
The cumulated risks observed at 70 years for breast cancer (77%) and for thyroid cancer (38%) are much higher than those observed in the general population (12% and <1%, respectively) and those described until recently in the literature (see online supplementary table S2).1 It is possible that these high estimates may be related to a recruitment bias in our study, because one third of the patients in this study presented with, or had already had, a cancer at the time of the request for PTEN gene analysis. However, most patients (2/3) did not have cancer when the PTEN gene analysis was requested. Further, one third of these patients were selected from genetic tests after identification of a mutation in the family and therefore did not present any recruitment bias. Interestingly, the cumulative risk of cancer is similar in this population to that in patients recruited for primary PTEN analysis (see online supplementary figure S7). In this respect, the cancer risks that we report appear not to be greatly overestimated, given the indications of the genetic analysis.
In addition, the prevalences of breast and thyroid cancers are similar to those reported in two recent articles presenting the same recruitment bias. The first study14 is a meta-analysis describing 211 CS patients, half (48%) of whom did not have deleterious germline PTEN mutations, and reporting a cumulated risk at 70 years of 81% for breast cancer and 21% for thyroid cancer. The second study13 describes 368 patients with deleterious germline PTEN mutations and reports cumulative risks at 70 years of 85.2% for breast cancer, 35.2% for thyroid cancer, and high SIRs of 25.4 and 51.1, respectively.
In addition to breast and thyroid cancer, four other tumour sites were overrepresented in this article.13 High SIR and cumulative risks at 70 years were reported for endometrium cancer (42.9% and 28.2%, respectively), renal cell cancer (30.6% and 33.6%), colorectal cancer (10.3% and 9%), and melanoma (8.5% and 6%). In our study, we observed high SIR for these sites compared to the general population, but the number of cases observed for endometrium cancer (4%, 3/70) and renal cancer (2%, 3/146) are lower than those reported by Tan et al.13 The risk of endometrium cancer may have been under-evaluated in our study because women with CS are often more likely to receive a hysterectomy for benign utero-ovarian tumours and are thus protected against the risk of cancer to the endometrium. Further, our results are not significant for kidney cancer in men and colorectal cancer. The risk of colorectal cancer is lower than the risk observed in other sites in contrast to the results from a recent series reporting a cumulative risk of 13% with a high SIR of 224.1 (95% CI 109.3 to 411.3).22 For melanoma, our results are significant for women and men and are associated with a higher number of cases in our study (6%) than in Tan et al's (2%). The melanoma risk for patients with PTEN mutations appears higher than previously reported, and with particularly early onset as the youngest case was for a 6-year-old patient in our series and a 3-year-old in Tan et al's study.
Overall, the risk of malignant tumours across all locations is very high, with a cumulative risk at 70 years of 85%. The cumulative risk at age 30 is also comparatively high at 23%. Cancer risk is higher for women in general (81%) than for men (48%), which is partially explained by the rate of breast cancer which is the most frequent cancer in CS. Interestingly, this observation contrasts with our observations for thyroid cancer with a higher SIR for men (199.5) than for women (43.2), supporting a previous report by Ngeow et al.21 This difference may be explained by the fact that, in a general population, thyroid cancer occurs less frequently in men. This does not appear to be the case for CS where we see similar rates of thyroid cancer for both men and women, suggesting that there may be a different determinism for thyroid cancer according to whether it is observed in the general population or in a CS population.
Genotype–phenotype correlations
We also investigated individual variations in cancer risk through an analysis of associations with different types of mutations, but no significant genotype–phenotype correlations were found. A significant correlation has been observed between truncating mutations and colorectal cancer,13 but we did not observe the same results, possibly because of such low numbers of colorectal cancer cases. Tan et al13 also found a significant correlation between mutations in the promoter and breast cancer; a finding that we did not seek to replicate as we did not analyse the PTEN promoter due to the debatable pathogenicity of these mutations.
Animal models have also reported a genotype–phenotype effect for two missense mutations (C124R and G129E) of the PTEN catalytic site and a loss of function mutation (PtenΔ4-5).23 The G129E mutation in mice appears to be frequently associated with an aggressive and varied tumoral phenotype, developing breast, thyroid and prostate tumours. Benign tumours of the endometrium are also described. The same tumoral spectrum (except for prostate) is observed for mice with C124R mutations, but the tumours are less frequent and less aggressive. These two missense mutations were not found in our series but have been described in the literature for patients with PHTS. A skin cancer and a breast cancer in one family, and a melanoma, a breast cancer and a glioblastoma in another family have been reported for four carriers of the G129E mutation.15 ,24 One case of colorectal cancer has also been described in one out of three families for three carriers of C124R mutations.15 ,25 Overall, half of the families with G129E mutations do not present any cancer. The patterns indicated from mice studies do not appear to be clearly reproducible in man.
Despite the description of some interesting phenotypes in mice and in man, no genotype–phenotype correlations appear to exist for CS. This impression is in agreement with previous reports15 ,17 and is supported by the large intra-familial variability in the disease, with the observation of different phenotypes such as CS, BRRS, LDD, SOLAMEN, macrocephaly-autism and juvenile polyposis syndromes for the same germline mutation. Several examples demonstrate this variability. One patient presented with a typical SOLAMEN syndrome while five other members of her family had a standard form of the disease.4 In another example in a family of four, the index patient was the only one to have LDD along with other signs of the disease; and in a final example in a family of five, the index patient was the only to present with a cognitive impairment. Overall, the absence of any demonstrated genotype–phenotype correlations indicates that the variability of the expression of the disease depends more on the action of modifying genes.26
Proposition of new surveillance recommendations
Finally, these updated cancer risks mean that adapting the care and management of patients with CS or PHTS is essential, especially for the risk of breast cancer for women, which appears similar to predisposition rates for BRCA1-2, but also for the risk of thyroid cancer where cases have been reported as early as age 10, 11 and 1327 ,28 and at 16 years in our series. New recommendations for the management of these patients are proposed in table 4. This management should vary according to the patient's age and sex.
Proposed surveillance recommendations for PTEN hamartoma tumour syndrome
In children, the assessment at diagnosis will primarily concern psychomotor development and research of dermatological, thyroid lesions and vascular malformations. Monitoring should be annual, including complete clinical examination, specific dermatologic examination, psychomotor assessment, and thyroid ultrasound from the age of 10 years.
In adults, the management is dominated by cancer risk. (1) The assessment at diagnosis must include a complete (and especially dermatological and neurological) clinical examination, mammography and breast MRI, thyroid ultrasound, transvaginal ultrasound, upper gastrointestinal endoscopy, colonoscopy, and renal ultrasound. A systematic brain MRI is more questionable because surgical treatment of brain lesions is indicated only if they are symptomatic. (2) The management of breast cancer risk depends on the quality of screening. If an important mammary dystrophy exists, annual clinical examination and breast MRI at the age of 20 years is recommended, or sooner if symptoms develop, and a prophylactic mastectomy can be discussed from the age of 25–30 years. In the case of moderate breast lesions, annual clinical examination and breast ultrasound are recommended from the age of 25 years, then annual mammography and breast MRI from the age of 30 years. (3) The management of endometrial cancer risk is based on an annual transvaginal ultrasound from the age of 35–40 years, and the indication for hysterectomy depends on the existence of benign lesions such uterine leiomyoma. (4) The management of thyroid cancer risk is based on an annual thyroid ultrasound and total thyroidectomy in cases of multiple nodules or multinodular goitre. (5) The management of kidney cancer risk is based on an annual renal ultrasound and/or renal MRI if there is a family history of renal cancer, or every 2 years if not. (6) The management of colorectal cancer risk is based on a systematic colonoscopy starting at age 30 years, then every 5 years or every 3 years if a severe polyposis and/or adenomatous polyps are found.
For the treatment of PHTS, three points are important: (1) complications of deep vascular malformations are frequent and justify an early treatment by embolisation or surgical resection; (2) the existence of melanoma in a child justifies excision of all suspicious melanic lesions; and (3) the existence of thyroid cancer after subtotal thyroidectomy justifies a total thyroidectomy when surgical resection of the thyroid lesions is indicated.
Acknowledgments
We thank the patients who contributed to this study. We thank Bernadette Gastaldello and Delfine Lafont who performed PTEN mutation analysis and Pippa McKelvie-Sebileau of Institut Bergonié for medical editorial assistance.
APPENDIX 1
The French Cowden Disease Network contributing members are: Marc Abramowicz, Medical Genetics Unit, Erasme Hospital, Brussels, Belgium; Didier Bessis, Dermatology Department, CHU, Montpellier, France; Eric Bieth, Medical Genetics Unit, CHU, Toulouse, France; Valérie Bonadona, Cancer Genetics Unit, Centre Léon Bérard, Lyon, France; Jean-Marie Bonnetblanc, Dermatology Department, CHU, Limoges, France; Liliane Demange, Cancer Genetics Unit, Institut Curie, Saint-Cloud, France; François Feillet, Paediatrics Department, Hôpital d'Enfants de Brabois, Vandoeuvre lès Nancy, France; Thierry Frebourg, Medical Genetics Department, CHU Charles-Nicolle, Rouen, France; Sophie Giraud, Medical Genetics Unit, Hôpital Edouard Herriot, CHU, Lyon, France; Irina Giurgea, Genetics Lab, CHIC, Créteil, France; Delphine Heron, Medical Genetics Unit, Hôpital la Pitié Salpêtrière, Paris, France; Muriel Holder, Medical Genetics Department, CHRU, Lille, France; Hubert Journel, Medical Genetics Unit, Centre Hospitalier Bretagne Atlantique, Vannes, France; Sophie Julia, Medical Genetics Department, CHU, Toulouse, France; Maha Kacem, Internal Medicine and Nephrology Unit, CHU Fattouma Bourguiba, Monastir, Tunisia; Sophie Lejeune, Medical Genetics Department, CHRU, Lille, France, Frédéric Leprat, Internal Medicine Unit, Polyclinique du Tondu, Bordeaux, France; Dominique Leroux, Medical Genetics Department, CHU, Grenoble, France; Catherine Lok, Dermatology Department, CHU, Amiens, France; Alain Lortholary, Cancer Genetics Unit, Centre Catherine de Sienne, Nantes, France; Stanislas, Lyonnet, Medical Genetics Department, Hôpital Necker-Enfants Malades, Paris, France; Geneviève Margueritte, Cancer Paediatrics Unit, CHU, Montpellier, France; Jacques Mauillon, Medical Genetics Department, CHU Charles-Nicolle, Rouen, France; Juliette Mazereeuw-Hautier, Dermatology Department, CHU, Toulouse, France; Sylvie Odent, Medical Genetics Department, CHU, Rennes, France; Clotilde Penet, Cancer Genetics Unit, Centre Jean Godinot, Reims, France; Anne Philippe, INSERM U781, Hôpital Necker-Enfants Malades, Paris, France; Henri Plauchu, Medical Genetics Department, CHU, Lyon, France; Ghislaine Plessis, Medical Genetics Unit, CHU Clémenceau, Caen, France; Emmanuel Plouvier, Paediatrics Department, CHU, Besançon, France; Marie-Aleth Richard, Dermatology Department, Hôpital Sainte Marguerite, CHU, Marseille, France; Abdelkrim Saadi, Department of Neurology, Hôpital de Benaknoun, Alger, Algeria; Jean-Christophe Saurin, Gastro-enterology Department, CHU, Lyon, France; Julie Tinat, Medical Genetics Department, CHU Charles-Nicolle, Rouen, France; Pierre Vabres, Dermatology Department, CHU, Dijon, France; Lionel Van Maldergem, Medical Genetics Unit, CHIC, Créteil, France; Philippe Vennin, Cancer Genetics Unit, Centre Oscar Lambret, Lille, France; Pierre-Jean Weiller, Internal Medicine Department, CHU, Marseille, France.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figures
- Data supplement 2 - Online tables
Footnotes
↵*A full list of the French Cowden disease network members is listed in appendix 1.
-
Contributors ML planned and supervised the work; VBu, EB, AD, PE, AB, VL, OC, BG, CDe, CDu, JPF, DB, ML and FC provided patient information and samples; VBr and SH performed statistical analyses; VBu analysed and interpreted the data; VBu, ML and FC wrote the manuscript. All authors revised and approved the final version of the manuscript.
-
Funding This work was supported by the Institut National du Cancer (INCa, Boulogne-Billancourt) as part of the French Cowden Syndrome Network.
-
Competing interests None.
-
Patient consent Obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed.