


No. Patients diagnosed with primary pulmonary coccidioidomycosis (PPC) who are asymptomatic or mildly symptomatic do not require treatment and can be monitored closely. Treatment should be initiated in patients with severe disease, extrathoracic dissemination, or risk factors such as immunosuppression.
WHEN SHOULD WE SUSPECT PRIMARY PULMONARY COCCIDIOIDOMYCOSIS?
Coccidioidomycosis, known colloquially as “valley fever,” is a fungal infection caused by the dimorphic fungus Coccidioides.1 It is endemic to the southwestern United States (Arizona and California) and northwestern Mexico. In fact, up to 29% of community-acquired pneumonia cases in Arizona are secondary to PPC.2 More recently, eastern Washington state has also been recognized as an endemic region.3
Inhalation of coccidioidal spores from dust can result in this illness, and the symptoms are clinically indistinguishable from those of community-acquired pneumonia.4 The most common presenting symptoms include fatigue, cough, headache, and night sweats.5 Patients may also have various systemic or rheumatologic complaints. It is important to note, however, that approximately 60% of patients with PPC are asymptomatic, and symptomatic patients often have self-limited disease.6 PPC should be suspected in individuals with an appropriate travel history who present with symptoms of pneumonia, and further laboratory testing should be undertaken to confirm the diagnosis.
HOW IS PRIMARY PULMONARY COCCIDIOIDOMYCOSIS DIAGNOSED?
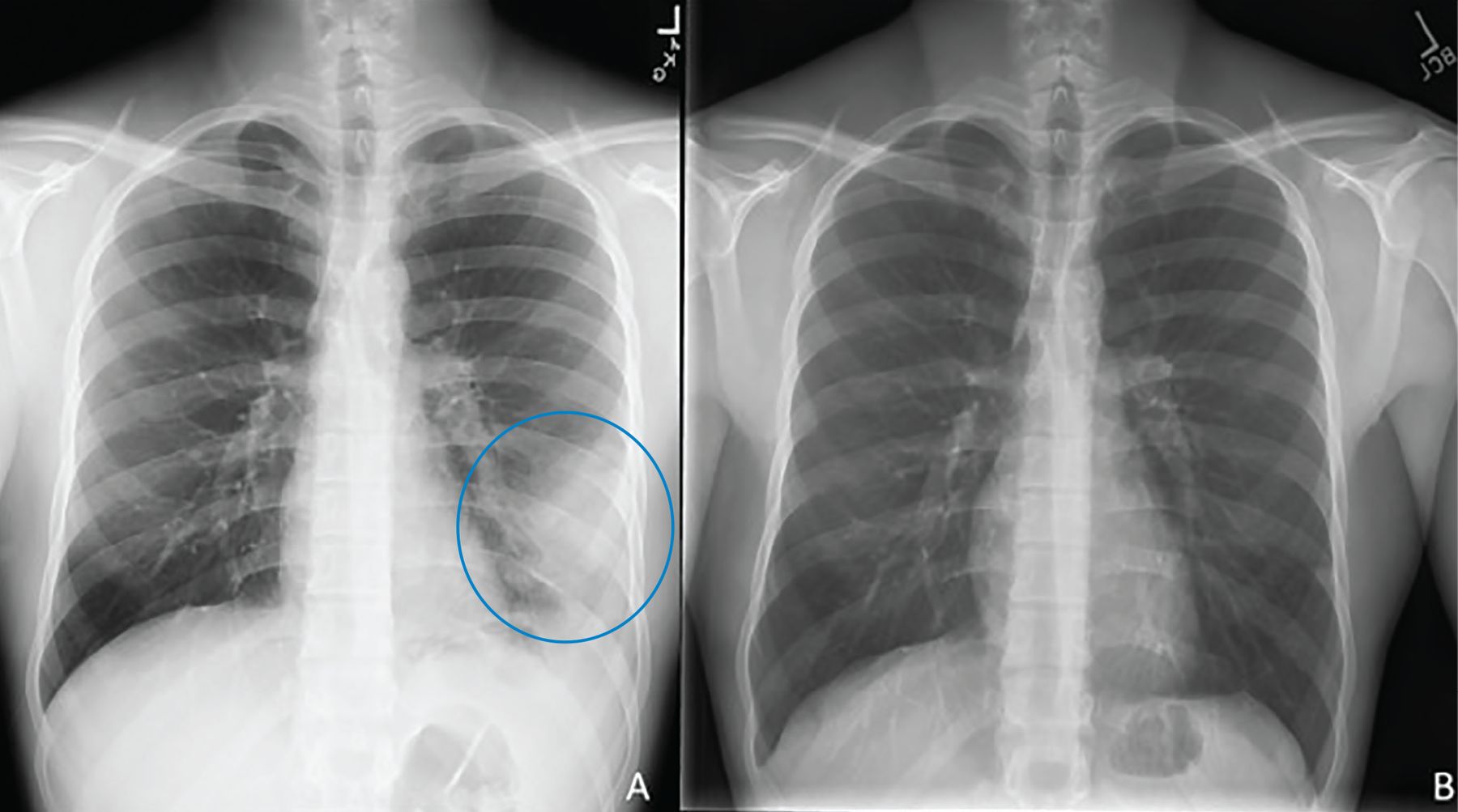
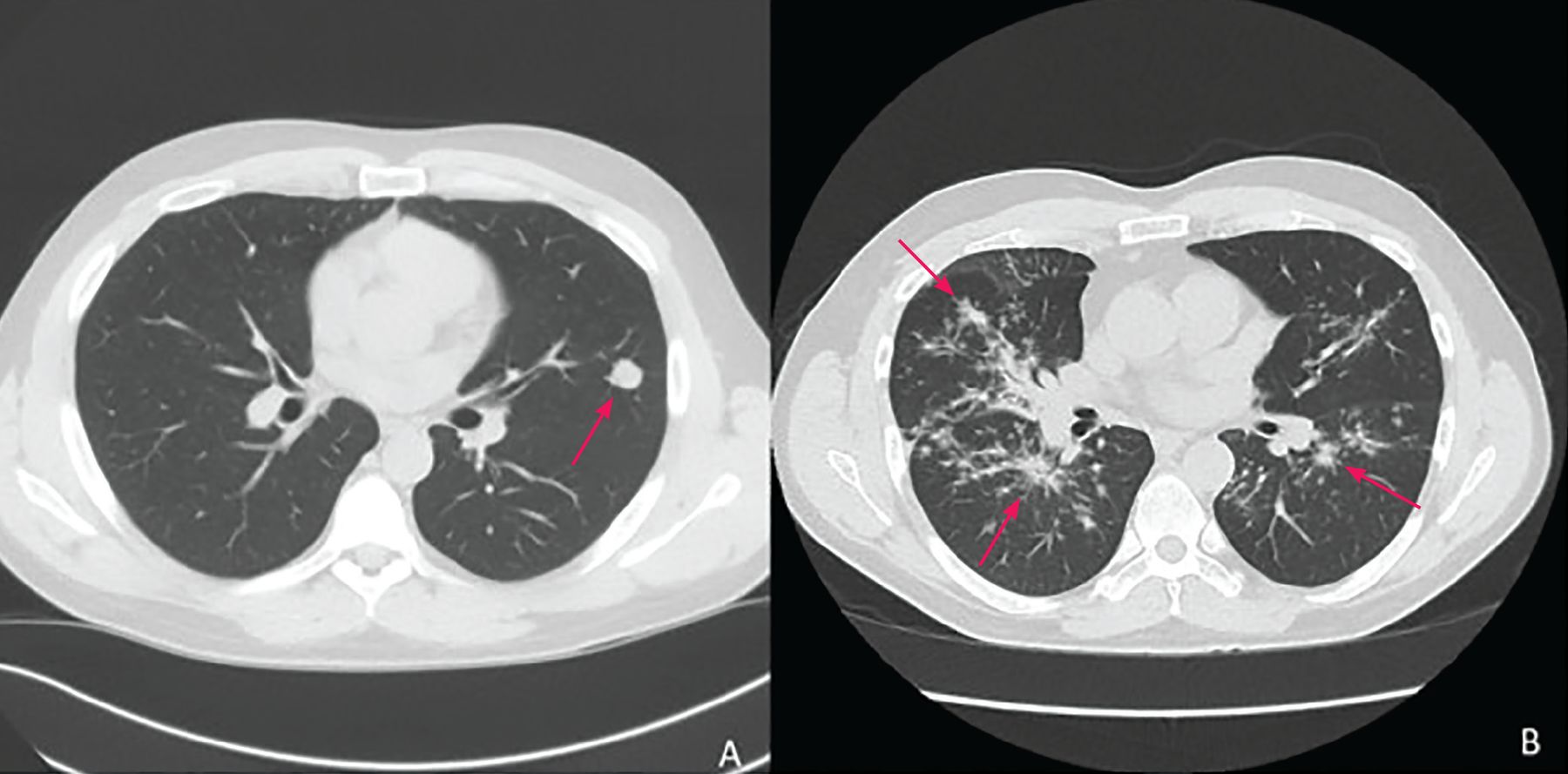
Diagnosis begins with clinical suspicion and an appropriate exposure history, along with clinical, radiographic, and laboratory features suggestive of PPC. Typical symptoms resemble pneumonia or bronchitis and are hard to distinguish from other bacterial and viral infections. Radiologic features (Figure 1, Figure 2) vary from pulmonary infiltrates (in most patients) to less common findings of pulmonary nodules or cavities, pleural effusions, and “tree-in-bud” changes.7
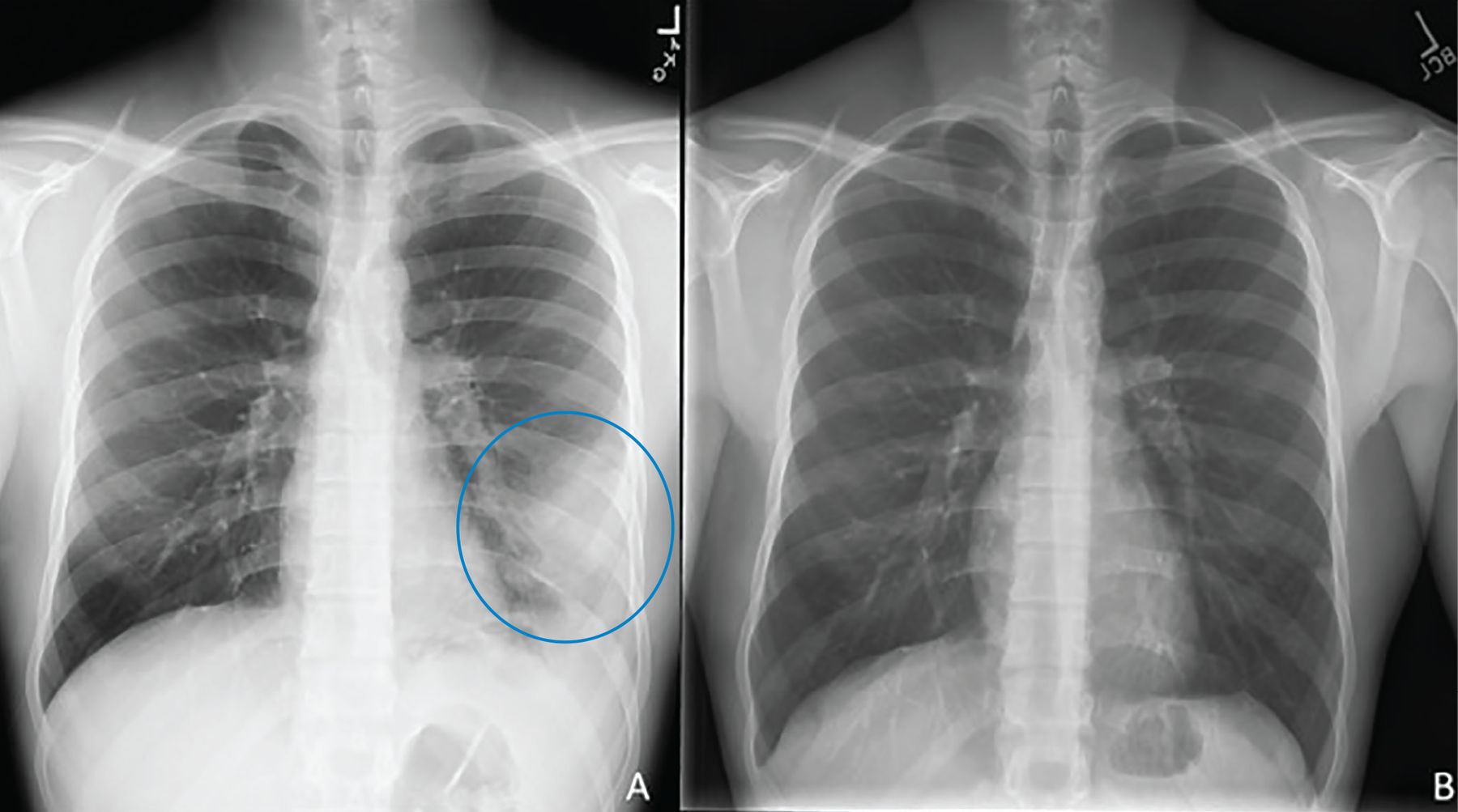
(A) A 22-year-old male with a left lower lung infiltrate (circle). (B) Repeat imaging 8 months after initiation of treatment showed interval clearance of previously visualized opacity.
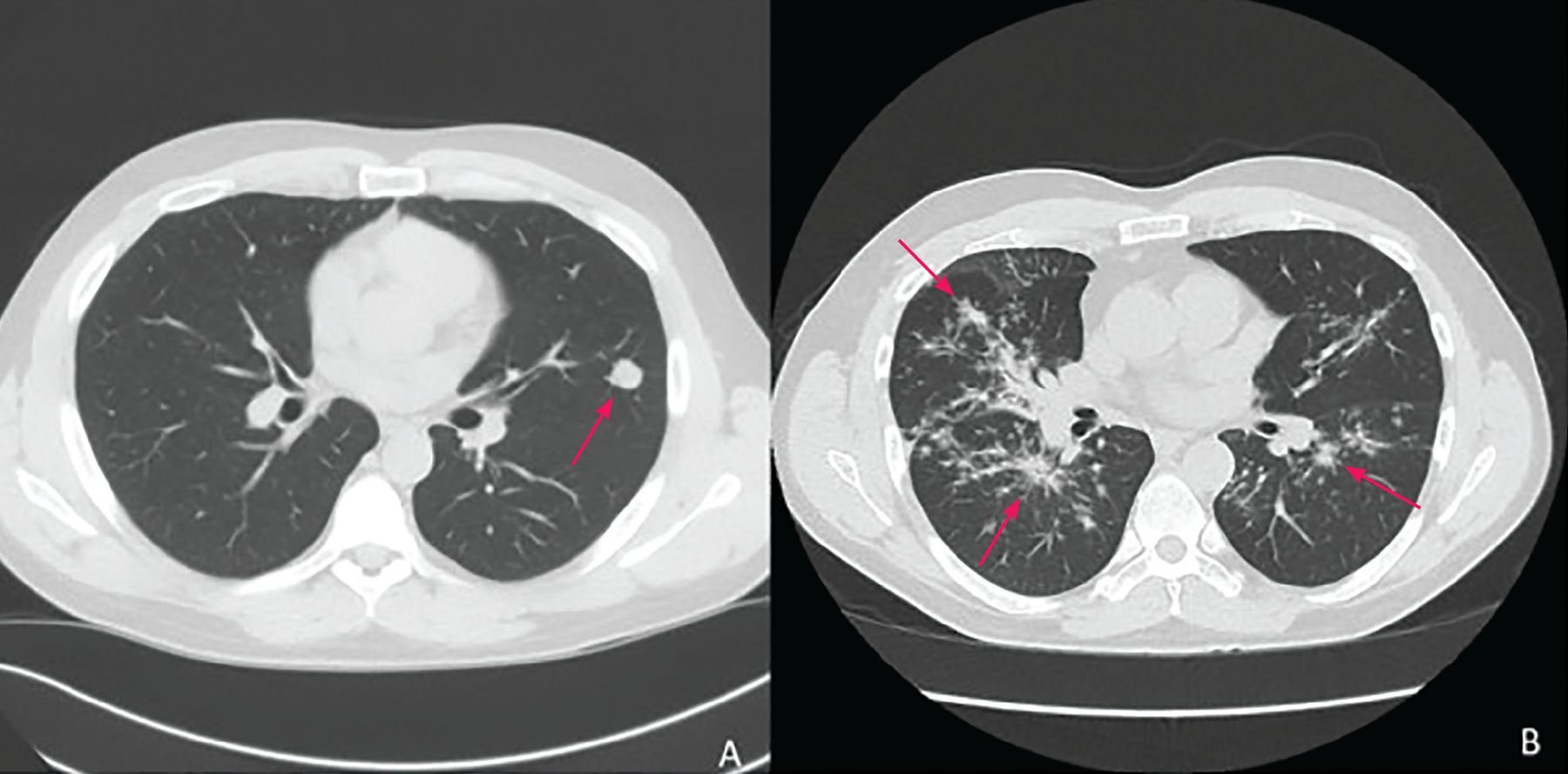
(A) A 1.5-cm lingular nodule with adjacent satellite nodularity (arrow) likely a noncalcified granuloma related to coccidioidomycosis. (B) Bilateral bronchovascular and perilymphatic nodules (arrows) seen in all lung fields, with subsequent bronchoalveolar lavage studies growing Coccidioides spp.
If sputum is available, culture provides a proven diagnosis since there is no state of colonization. However, most patients manifest a dry cough, and serologic tests are most commonly used for diagnosis.8 These include enzyme immunoassay (EIA), immuno diffusion, and complement fixation testing to detect immunoglobulin (Ig) M and IgG antibodies. Maximal sensitivity for diagnosis of coccidioidomycosis occurs with positive test results for both IgM and IgG by EIA (Figure 3).7 An isolated positive IgM by EIA is often a false-positive and thus requires either repeat testing to demonstrate seroconversion or subsequent microbiologic, cytologic, or histopathological testing from tissue biopsy or body fluid (eg, bronchoalveolar lavage). An isolated positive IgG by EIA antibody titer is typically confirmed by further testing with immunodiffusion and complement fixation IgG and IgM. Complement fixation IgG provides a baseline quantitative titer that can be followed over time. However, the turnaround time for immunodiffusion and complement fixation testing is long because these tests often have to be sent to a reference laboratory.1,7 Sensitivity of immunodiffusion is approximately 73%, and for complement fixation approximately 75%.7 In the case of extrathoracic coccidioidomycosis (ie, involvement of skin and soft tissue, bone and joint, or meninges), especially in immunosuppressed patients, testing for serum or urine antigen may also be useful.7

Algorithm for serologic testing in suspected coccidioides infection.
a Suspected in the presence of typical symptoms and radiographic findings.
b Definitive diagnosis requires positive biopsy, tissue culture, or body fluid culture.
CF = complement fixation testing; ID = immunodiffusion testing; IgG = immunoglobulin G; IgM = immunoglobulin M
TREATMENT OPTIONS
The decision to treat PPC should be individualized, since most patients will not require antifungal treatment (Table 1). The current Infectious Diseases Society of America (IDSA) guidelines recommend patient education, close observation, and supportive measures such as a reconditioning physical therapy program for patients with mild symptoms, or for those who have significantly improved by the time of diagnosis.4 Treatment is recommended for patients with prolonged symptoms (for example, symptoms that persist for > 2 months or severe night sweats for > 3 weeks), extensive pulmonary involvement (eg, > 50% involvement of one or both lungs), or severe disease requiring hospitalization. Additionally, guidelines recommend treating patients with concurrent diabetes and those with underlying cellular immune deficiencies, such as transplant patients on antirejection therapy, persons with human immunodeficiency virus infection with CD4 counts below 250, and patients on high-dose corticosteroids. Treatment can be considered for patients of African or Filipino descent.4
Situations in which to consider antifungal therapy
No randomized trials have been conducted to assess whether treatment of uncomplicated coccidioidal infection improves time to symptom-free period or prevents progression of disease. However, experts have observed benefit in treatment of patients with severe disease. Expert opinion varies, but IDSA guidelines suggest that severe disease can be considered when one or more of the following are present: weight loss greater than 10%, intense night sweats for more than 3 weeks, involvement of more than half of one lung or of both lungs, significant adenopathy, antibody titers greater than 1:16, symptoms for longer than 2 months, or inability to work.4
If initiated, treatment of mild to moderate PPC should begin with an orally absorbed azole such as fluconazole at a daily dose of at least 400 mg, for approximately 3 to 6 months or as driven by illness course. It is important to counsel patients on possible fluconazole-related adverse effects such as gastrointestinal upset, frequent cheilitis, reversible alopecia, and skin and nail changes, as these effects could result in medication nonadherence. Itraconazole is another first-line option, but fluconazole is usually preferred due to its lower cost, fewer drug interactions, and better patient tolerance.
In patients whose PPC is rapidly progressing—as evidenced clinically by signs such as need for hospitalization, worsening mental status, or increasing oxygen requirements—IDSA guidelines recommend consideration of liposomal amphotericin B.4 With biopsy-proven extrapulmonary disseminated disease, higher doses of fluconazole (such as 800 mg daily) should be considered. Patients failing to improve on fluconazole or itraconazole should be considered for higher-generation azole therapy such as posaconazole. Infectious disease consultation for these patients should be considered.
EXPERT OPINION ON MONITORING OF PATIENTS WITH PPC
Patients should be followed regularly for improvement in clinical symptoms, serology, and radiographic findings in order to monitor for disease complications. These include symptomatic cavitary lung lesions accompanied by secondary bacterial infection, pain, or hemoptysis (hemoptysis would require consideration of surgical excision), or dissemination to the meninges, skin, or bone and joint.
Patients can initially be seen in the office at least every 4 to 12 weeks, depending on how ill they are, and this monitoring can be extended to every 6 months as symptoms improve. Complement fixation testing for anticoccidioidal antibodies should also be repeated every 12 weeks, even with clinical improvement, to ensure titers decrease, as an increase in titers could be a sign of treatment failure or progression to extrapulmonary dissemination. Titers greater than 1:32 may suggest continued fungal growth or refractory disease, and changes in treatment could be considered.9 Similarly, if there is evidence of abnormal imaging on initial evaluation, this should be checked again at approximately 12 weeks and again several months later to monitor for residual disease or resolution. Serum transaminase levels should be monitored initially and then periodically, as antifungal therapy has been associated with hepatocellular injury.10
SPECIAL CONSIDERATIONS IN PREGNANCY
Pregnant patients with nonsevere disease can be monitored as other immunocompetent patients. Azole therapy has been associated with increased rates of spontaneous abortion and birth defects in infants, thus warranting a US Food and Drug Administration warning.6 Thus, azoles should be avoided when possible during the first trimester. Amphotericin is effective for pregnant patients and safe for the fetus but has multiple known adverse effects, including effects on the kidneys and electrolytes of the patient. Because of the potential for both severe coccidioidomycosis and harms from treatment, an infectious disease consultation is reasonable.
THE BOTTOM LINE
PPC is a fungal infection found most commonly in the southwestern United States and northern Mexico, but outbreaks of travel-associated coccidioidomycosis have been identified around the world. PPC should be considered in the differential diagnosis of patients with recent travel to endemic areas who present with symptoms of pneumonia and bronchitis.
Most patients do not require treatment, but the decision to treat should be individualized and based on a variety of factors. First-line antifungal treatment consists of fluconazole at least 400 mg daily, with certain exceptions such as avoiding its use in the first trimester of pregnancy. Whether treatment is or is not provided, close follow-up of all patients is recommended.
DISCLOSURES
The authors report no relevant financial relationships which, in the context of their contributions, could be perceived as a potential conflict of interest.
- Copyright © 2022 The Cleveland Clinic Foundation. All Rights Reserved.
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.