


ABSTRACT
Diabetic ketoacidosis (DKA) was historically considered a condition typical of type 1 diabetes. However, patients with type 2 diabetes may present with DKA, usually with higher blood glucose levels and milder ketoacidosis. With the increased use of sodium-glucose cotransporter 2 (SGLT-2) inhibitors, the variant euglycemic DKA has been described. SGLT-2 inhibitors cause a low level of ambient ketones; any additional ketone formation predisposes to ketoacidosis, while the agent’s glycosuric effect limits hyperglycemia. The principles of DKA management are fluid administration, electrolyte control, and glucose control with insulin. In euglycemic DKA, the immediate use of a glucose-containing intravenous fluid induces endogenous insulin secretion and stops ketogenesis. Due to the half-life of SGLT-2 inhibitors, the duration of euglycemic DKA may be more prolonged.
Management of DKA requires strict discipline and a hierarchal approach—fluid administration; electrolyte and, especially, potassium control; and hyperglycemia therapy.
When ambient glucose levels are less than 200 to 250 mg/dL at presentation or during treatment, or if there is a history of SGLT-2 inhibitor therapy, the initial intravenous fluid must contain dextrose.
Before starting or resuming an SGLT-2 inhibitor, an in-depth evaluation of euglycemic DKA risks and an explanation of how to mitigate them are essential.
Ketoacidosis is a ubiquitous condition usually specified by its etiology. Thus, the forms of ketoacidosis—starvation, alcoholic, hyperemesis gravidarum, and diabetic—all have a common metabolic pathway to ketoacidosis: the inability to metabolize glucose as a primary fuel, setting into motion the production of secondary fuels that generate strongly acidic ketone bodies: acetoacetate, beta hydroxybutyrate, and acetone. That inability may stem from a lack of available glucose, like in starvation, or a decrease in hepatic gluconeogenesis induced by alcohol, leading to decreased insulin secretion, increased lipolysis, impaired shunting of fatty acids to mitochondria, fatty acid oxidation, and subsequent ketogenesis, causing an elevated anion-gap metabolic acidosis. In hyperemesis gravidarum, the lack of ingested glucose and the resultant decrease in insulin are amplified by the presence of insulin resistance during pregnancy. In these circumstances, the ambient glucose level is most frequently in the low-normal range, and mild hyperglycemia is rare.
Diabetic ketoacidosis (DKA), on the other hand, is defined by a triad of hyperglycemia with an ambient blood glucose level well greater than 250 mg/dL, high anion-gap acidosis, and increased plasma ketones, which are the unmeasured anions causing the gap.1 Euglycemic DKA (EDKA) is a variant of DKA in which the blood glucose is less than 250 mg/dL but the other features of DKA are present. Although EDKA is not common, it can occur in type 1 diabetes and has become more frequent in type 2 diabetes, especially with the growing use of sodium-glucose cotransporter 2 (SGLT-2) inhibitors.
|
SGLT receptors are expressed by islet alpha cells. However, although SGLT-1 receptors have been described, evidence supporting the presence of SGLT-2 receptors in alpha cells is inconsistent, and cross-reactivity of SGLT-2 inhibitors on the SGLT-1 receptor is unlikely.2 Paracrine effects on the secretion of glucagonotropic and static substances from other islet cells have been proposed as an alternative mechanism, but this hypothesis has not been conclusively proven. Of interest is the presence of a renal–pancreatic loop, which suggests that glycosuria caused by the SGLT-2 inhibitor and the associated lowering of circulating glucose triggers the increase in glucagon. This hypothesis is supported by data from glucose clamping experiments in which the rise in glucagon was prevented if the blood glucose level did not change during SGLT-2 inhibitor therapy.2 |
This article reviews the disease process of diabetic ketoacidosis, what to consider before starting patients on SGLT-2 inhibitors, and the differences in the approach to managing DKA and EDKA.
PATHOPHYSIOLOGY
Ketoacidosis in diabetes
When there is an absolute deficiency of insulin (as in type 1 diabetes) or severely augmented insulin resistance (by elevation of stress hormones and inflammatory cytokines in individuals predisposed to insulin resistance), the normal suppressive effect of insulin on glucagon is no longer operative. The unrestrained rise in glucagon disrupts the insulin-to-glucagon ratio in favor of glucagon, resulting in enhanced gluconeogenesis and release of free fatty acids. Fatty acid oxidation results in the formation of ketone bodies, which are secondary fuels used by organs and tissues such as the heart and muscles. In these tissues, beta hydroxybutyrate is converted back to acetoacetate, which is a substrate for the tricarboxylic acid (or Krebs) cycle that synthesizes and metabolizes acetyl-coenzyme A to yield adenosine triphosphate and carbon dioxide.
The rate-limiting steps in the ongoing metabolism of ketone bodies are nicotinamide adenine dinucleotide and its metabolites and succinyl coenzyme A. When these steps are overwhelmed, ketones accumulate and, because they are strong acids, induce acidosis. At the same time, the lack of insulin does not allow for the removal of glucose from the bloodstream, causing hyperglycemia and leading to osmotic diuresis, which promotes dehydration.
The EDKA variant
EDKA due to SGLT-2 inhibitors has a somewhat different etiologic basis: SGLT-2 inhibitors intrinsically stimulate the release of glucagon.2 The mechanism is unclear; the sidebar “Glucagon release in EDKA: Plausible pathophysiologic mechanisms” outlines the proposed mechanisms.
The rise in glucagon changes the glucagon-to-insulin ratio and ensures that treatment with SGLT-2 inhibitors fosters a higher level of circulating ketones in the steady non-EDKA state.3 Any additional reasons for developing ketones, such as keto diets or “nothing by mouth after midnight” (who isn’t?) and prolonged fasting, can tip the balance into ketoacidosis. The effect of SGLT-2 inhibitors on glycosuria continues, limiting hyperglycemia while promoting dehydration.
Notably, the half-life of SGLT-2 inhibitors ranges between 13 and 17 hours, so their effect can persist for up to 3 days (4.5 half-lives), prolonging the duration of EDKA and the treatment regimen. In addition, SGLT-2 inhibitor–induced glycosuria is accompanied by natriuresis, which disrupts the electrochemical gradient in the tubular fluid and drives the reabsorption of negatively charged ketone bodies,4 further contributing to ketoacidosis.
CONSIDER RISK FACTORS
Table 1 shows the risk factors that need to be mitigated before starting a patient on an SGLT-2 inhibitor. In instances of significant defects in insulin secretion, basal insulin administered exogenously at an adequate dose negates the risk of EDKA. Creatinine is a byproduct of muscle metabolism; thus, a low value is highly suggestive of sarcopenia. Muscles use ketones as a fuel, and a person with low muscle mass will not use as much, allowing for a greater accumulation of ketones. It is crucial to establish and treat the cause of preexisting acidosis.
Potential higher risk factors of euglycemic diabetic ketoacidosis and their mechanisms
SGLT-2 inhibitors protect renal function, but their use in chronic kidney disease needs extra care because they can predispose individuals to dehydration, thereby precipitating an acute renal injury (in which case the agent must be discontinued at least temporarily). The need to maintain adequate hydration must always be stressed to patients starting an SGLT-2 inhibitor.
Factors predisposing to EDKA
As outlined previously, SGLT-2 inhibitor therapy is always accompanied by an increase in ketonemia. Thus, any circumstances that would normally cause ketogenesis will further enhance ketonemia and may lead to ketoacidosis. These factors, which need to be reviewed with the patient, are as follows:
Prolonged fasting (pre- and postoperatively)
Ketogenic diets
Anorexia and bulimia
Alcohol intoxication
Insulin pump malfunction
Gastroenteritis and pancreatitis
Gastroparesis.
In the event of an emergency that necessitates fasting and the patient has not been able to discontinue the SGLT-2 inhibitor, an intravenous drip with dextrose (glucose 4–5 g per hour) to offset starvation and either a basal insulin dose or an intravenous insulin infusion, depending on the clinical status of the patient, must be started to induce a favorable insulin-to-glucagon ratio and to stop ketogenesis. There are no studies on the appropriate dose; however, in our experience, basal insulin 0.15 to 0.25 units/kg, without exceeding 20 to 25 units, is adequate to minimize risk of both EDKA and hypoglycemia.
PATIENT EDUCATION BEFORE STARTING SGLT-2 INHIBITORS
It is important to discuss the benefits and risks of SGLT-2 inhibitors with patients and to also outline some crucial dos and don’ts.
Dos
Maintain hydration with water and a small amount of electrolyte solution
Ensure 30% to 35% carbohydrate content in all meals and snacks
Limit alcohol intake
Know the identifying symptoms: fruity breath, thirst, polyuria and nocturia, nausea and vomiting, abdominal pain, confusion, and fever.
Don’ts
If on basal insulin and self-managing, do not decrease the dose more than 20% without discussion with the supervising healthcare team
No keto diets
Do not take the SGLT-2 inhibitor starting 3 days before fasting longer than overnight.
Sick-day rules
In acute intercurrent illness with nausea, vomiting, or diarrhea, sip on calorie-dense electrolyte solutions (200 mL every 30 minutes), suck on hard candy, or consume a tablespoon of sugar every 15 to 20 minutes. If there is no improvement and symptoms persist, stop the SGLT-2 inhibitor and go to an emergency department or call the primary care clinician. Once in the emergency department, inform medical staff about the need to start a glucose-containing intravenous infusion.
MANAGEMENT PRINCIPLES
Regardless of cause, management of ketoacidosis requires strict discipline and, at least at the start, a hierarchal approach.
Fluids
The primary goal of fluid administration is to restore tissue perfusion. The standard requirement is 15 to 20 mL/kg in the first hour, which is about 1 to 1.5 L.1 Two priorities are addressed: dehydration and resolution of the component of hypernatremia caused by hemoconcentration before any significant electrolyte shifts happen between the intracellular and extracellular compartments. Normal saline remains the fluid of choice in the first hour, especially because the potassium level is initially not known and balanced electrolyte solutions contain potassium (albeit in low concentrations).
Fluid choice after the first bolus also depends on the corrected sodium level:
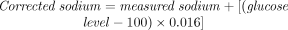
If the result reveals hyponatremia (serum sodium < 134 mmol/L), changing to a balanced electrolyte solution is helpful; however, in the face of normo- or hypernatremia, delaying the change to isotonic crystalloid solutions and using half normal saline may be more advisable.
The choice of which fluid is optimal after the bolus of normal saline and achieving an appropriate sodium level is open for discussion. The option of continuing normal saline vs changing to a balanced crystalloid solution such as Ringer’s lactate solution or a balanced electrolyte solution like Plasma-Lyte has been studied.5,6 The higher chloride and osmolarity of normal saline are associated with the development of hyperchloremic metabolic acidosis.5 Balanced crystalloids can be used for volume expansion and are not associated with hyperchloremic acidosis. In a subgroup analysis of 172 adults with DKA from 2 large cluster-randomized clinical trials, the median time to DKA resolution was significantly shorter with balanced crystalloids (13.0 hours) compared with saline (16.9 hours),5 and balanced crystalloids resulted in less hyperchloremia and a faster recovery of bicarbonate levels.6
When the circulating glucose level is less than 250 mg/dL, giving a 5% dextrose solution containing balanced electrolytes is necessary to prevent hypoglycemia and to reduce the risk of cerebral edema caused by rapid correction of the extracellular compartment.7
In EDKA, hydration must be started with a dextrose-containing fluid to accomplish 4 crucial goals:
Provide glucose to stop the ketogenic process
Reinstitute secretion of endogenous insulin, if present, thereby starting to alter the insulin-to-glucagon ratio in favor of insulin, stopping the ketogenic process
Rehydrate and replete solutes
Counterbalance the effects of the infused insulin that was started to enhance recovery from ketoacidosis.
There are no specific studies related to fluid replacement in EDKA. However, in EDKA due to SGLT-2 inhibitors, hypernatremia is less of an issue because SGLT-2 inhibitors cause glycosuria, which also induces natriuresis. Theoretically, the persistence of acidosis caused by the continuing effect of SGLT-2 inhibitors in EDKA may make the use of a balanced electrolyte fluid even more beneficial.
Potassium
The serum potassium value is not an indicator of potassium status in either DKA or EDKA because in each form there is a total potassium deficit due to osmotic diuresis. Volume expansion, correction of acidosis, and insulin therapy all lower potassium levels. Therefore, once serum potassium levels are less than 5.3 mmol/L, potassium replacement is essential.1 If hypokalemia is present at the time of diagnosis, potassium must be added to the initial fluid administered. Insulin should be started only after the serum potassium level is greater than 3.3 to 3.5 mmol/L to protect against arrhythmias, respiratory muscle weakness, and even death.
In SGLT-2 inhibitor–related EDKA, the SGLT-2 inhibitor generally lowers the risk of severe hyperkalemia in people with type 2 diabetes.8
Insulin therapy
Insulin therapy is optimally instituted after the first bolus of intravenous fluid. This delay allows for a more accurate estimation of hyperglycemia by correcting hemoconcentration and a more meaningful evaluation of the serum potassium level.
Intravenous insulin to treat DKA is preferable. Whereas hourly subcutaneous short-acting insulin analogs have been used successfully to treat mild to moderate DKA,9,10 the consensus remains with intravenous therapy. Studies on the utility of an initial bolus followed by an infusion have shown no beneficial effect of a bolus dose on the rate of glucose decrease, the rate of anion-gap correction, or the hospital length of stay.11 However, a bolus shows a trend toward more frequent development of hypoglycemia (6% vs 1%).
In EDKA, a simultaneous infusion of insulin and a dextrose-containing fluid is essential; insulin augments the effect of the infused glucose, as described above.
Bicarbonate
A systematic review of bicarbonate therapy in DKA showed adding bicarbonate produced a transient improvement in metabolic acidosis, but had no effect on glycemic improvement and also carried a risk of cerebral edema in children.12 Even when used in severe acidotic DKA, bicarbonate therapy did not show any difference in time to resolution of acidosis, length of time intravenous insulin was required, potassium supplementation requirements, or hospital length of stay.13
There are no studies on bicarbonate administration in severe acidosis (pH < 6.9).12 If pH is between 6.9 and 7.0 and bicarbonate therapy is being considered, 50 mmol of bicarbonate in 200 mL of sterile water with 10 mmol of potassium chloride can be given over 2 hours to achieve a pH greater than 7.0.1 Adding potassium chloride simultaneously is dependent on ambient potassium levels because administering bicarbonate may increase risk for hypokalemia.
The discussion regarding bicarbonate therapy in EDKA is even more germane because bicarbonate levels may remain low until the SGLT-2 inhibitor is completely cleared (up to 4.5 half-lives, which is approximately 60–72 hours). Accordingly, it may be tempting to increase the bicarbonate level with exogenous bicarbonate therapy, but there is no proven benefit.12,14
Phosphate
Hypophosphatemia during DKA is common and increases with severe acidosis. The increased loss of phosphate is a result of transcellular shift, osmotic diuresis, and reduced phosphate reabsorption in the renal proximal tubule due to acidosis and hyperglycemia. With administration of insulin and fluids, phosphate shifts into the intracellular compartment and a nadir of phosphate is reached at a median 16 hours into therapy.15 However, there appears to be no adverse effects from hypophosphatemia when left untreated, and no benefits from treatment have been observed.
There is a concern that very low phosphate levels (< 1 mg/dL) may contribute to cardiac and muscle weakness and respiratory depression. In such circumstances, potassium phosphate 20 to 30 mmol/L can be added to the replacement fluid to achieve a phosphate level just greater than 1 mg/dL.14 Overenthusiastic replacement not only shows no benefit, but also can result in significant hypocalcemia.
In EDKA from SGLT-2 inhibitors, low phosphate is uncommon because these agents usually raise phosphate levels by increasing renal tubular reabsorption of phosphate.16
Transitioning from intravenous insulin to subcutaneous insulin
The current recommendations are to start subcutaneous insulin when the serum glucose level is less than 200 to 250 mg/dL, the anion gap is less than 12, and the bicarbonate level is greater than 15 mmol/L.17 Under those circumstances, basal insulin needs to be administered at the preadmission dose (if known) or at 0.25 units/kg (if unknown) and should overlap with intravenous insulin for about 2 hours. Planning a transition without the use of basal insulin is harmful.
In addition, studies have shown benefits of early institution of basal insulin while using intravenous insulin to treat DKA, including less rebound hyperglycemia when intravenous insulin is discontinued, quicker resolution of DKA, reduced intravenous insulin requirements, and reduced hospital length of stay, with no increase in hypoglycemia or hypokalemia.17
In SGLT-2 inhibitor–related EDKA, the transition cutoffs need to be tempered because ketosis—and, therefore, a low bicarbonate level—may rebound after intravenous therapy is discontinued due to the persistent effect of the medication. Thus, it is important to ensure that the anion gap has normalized and the bicarbonate level is in the low-normal range before transitioning.
DO SGLT-2 INHIBITORS HAVE TO BE DISCONTINUED AFTER AN EPISODE OF EDKA?
A blanket no may be a medically and legally appropriate answer, but it should be noted that insulin is not discontinued even after serial episodes of DKA. The answer should be based on an in-depth review of the risk-benefit ratio and a discussion with the patient, as described above, because therapy must be individualized.
Before restarting the SGLT-2 inhibitor, a detailed review to discern the exact cause of the EDKA episode is essential. If the SGLT-2 inhibitor is restarted, the patient must be informed about the contributing factors that caused the DKA (fasting, surgery, and so on), so that, if faced with a similar situation in the future, the medication can be discontinued or early precautions started, as outlined above.
In relatively lean individuals with high hemoglobin A1c levels, there is a higher chance of insulin insufficiency. Thus, when restarting an SGLT-2 inhibitor, initiating basal insulin or, in rare circumstances, an insulin secretagogue like a sulfonylurea may be prudent.
Using an incretin, whether glucagon-like peptide-1 or dipeptidyl peptidase 4 inhibitor, in addition to an SGLT-2 inhibitor is not appropriate because these agents do not independently stimulate insulin release. Instead, they are dependent on ambient glucose to facilitate insulin release, so fasting and a falling glucose level will still deactivate insulin secretion while on these agents. There are reports of patients presenting with EDKA on combinations of incretins and SGLT-2 inhibitors. Development of EDKA is also possible while on glucagon-like peptide-1 receptor agonists18,19 and dipeptidyl peptidase 4 inhibitors.20 There are also reports of EDKA in patients without diabetes on SGLT-2 inhibitors for heart failure21 due to the underlying insulin resistance common to heart failure.
CONCLUSION
Although ketoacidosis has a common pathophysiologic pathway to development, varied etiopathologies initiate the process, chief among them being diabetes. With the extensive use of SGLT-2 inhibitors, it has become clear that the pathophysiology of DKA is different from that of EDKA (Table 2). Therefore, most importantly, the management strategies for DKA and EDKA are different, as are the surveillance requirements for terminating acute interventions. Given these differences, significant modifications in the clinical approach are needed.
Differences between diabetic ketoacidosis and euglycemic diabetic ketoacidosis
DISCLOSURES
Dr. Mehta has disclosed teaching and speaking for Novo Nordisk, Inc. Dr. Zimmerman has disclosed ownership interest (stock, stock options in a publicly owned company) in Baxter, research funding from Bayer, and teaching and speaking for Xeris.
Acknowledgments
The authors thank Dr. Charles Faiman for his invaluable help with formatting and editing this manuscript.
- Copyright © 2025 The Cleveland Clinic Foundation. All Rights Reserved.



