


ABSTRACT
The pathogenesis of immunoglobulin (Ig) A nephropathy is described through a “4-hit” model involving production of galactose-deficient IgA, production of autoantibodies to galactose-deficient IgA, and subsequent deposition of immune complexes in the kidney glomerulus. Diagnosis remains dependent on a kidney biopsy, often after hematuria or proteinuria is detected on urinalysis. The cornerstone of therapy still involves renin-angiotensin-aldosterone system inhibitors or corticosteroids; however, new therapies targeting key aspects of the pathogenesis of IgA nephropathy are being introduced.
IgA nephropathy is a relatively common autoimmune glomerular disease that can be diagnosed only by biopsy.
Proteinuria reduction remains the most important treatment target.
Treatment now includes sodium-glucose cotransporter 2 inhibitors and endothelin receptor antagonists in addition to renin-angiotensin-aldosterone system inhibitors and corticosteroids.
New therapies target multiple pathogenic “hits” to reduce proteinuria and preserve kidney function.
A Major cause of kidney failure in children and adults, immunoglobulin (Ig) A nephropathy is the most common primary glomerulonephritis; its worldwide incidence is at least 2.5 per 100,000.1
There has been a tremendous lag in the treatment of the disease since its histologic features were first described in 1968 by Berger and Hinglais.2 For decades, nephrologists have had little more than renin-angiotensin-aldosterone system (RAAS) inhibitors or corticosteroids in their treatment armamentarium. Thanks to a recent transformation in our understanding of and therapeutic approach to IgA nephropathy, in the near future, there may be more therapeutic options for IgA nephropathy than for any other glomerular disease. Opportunities for new therapies stem from the acknowledgment by the US Food and Drug Administration (FDA) that proteinuria reduction is an acceptable trial end point3 in the path to drug approval. This recent innovation is also a direct consequence of years of basic science research that has refined our understanding of the pathogenesis of IgA nephropathy into a framework of “4 hits,” with each hit representing a target of novel therapies.
This review addresses the current approach to management of IgA nephropathy and therapeutic options we can soon expect.
THE 4 HITS OF IgA NEPHROPATHY
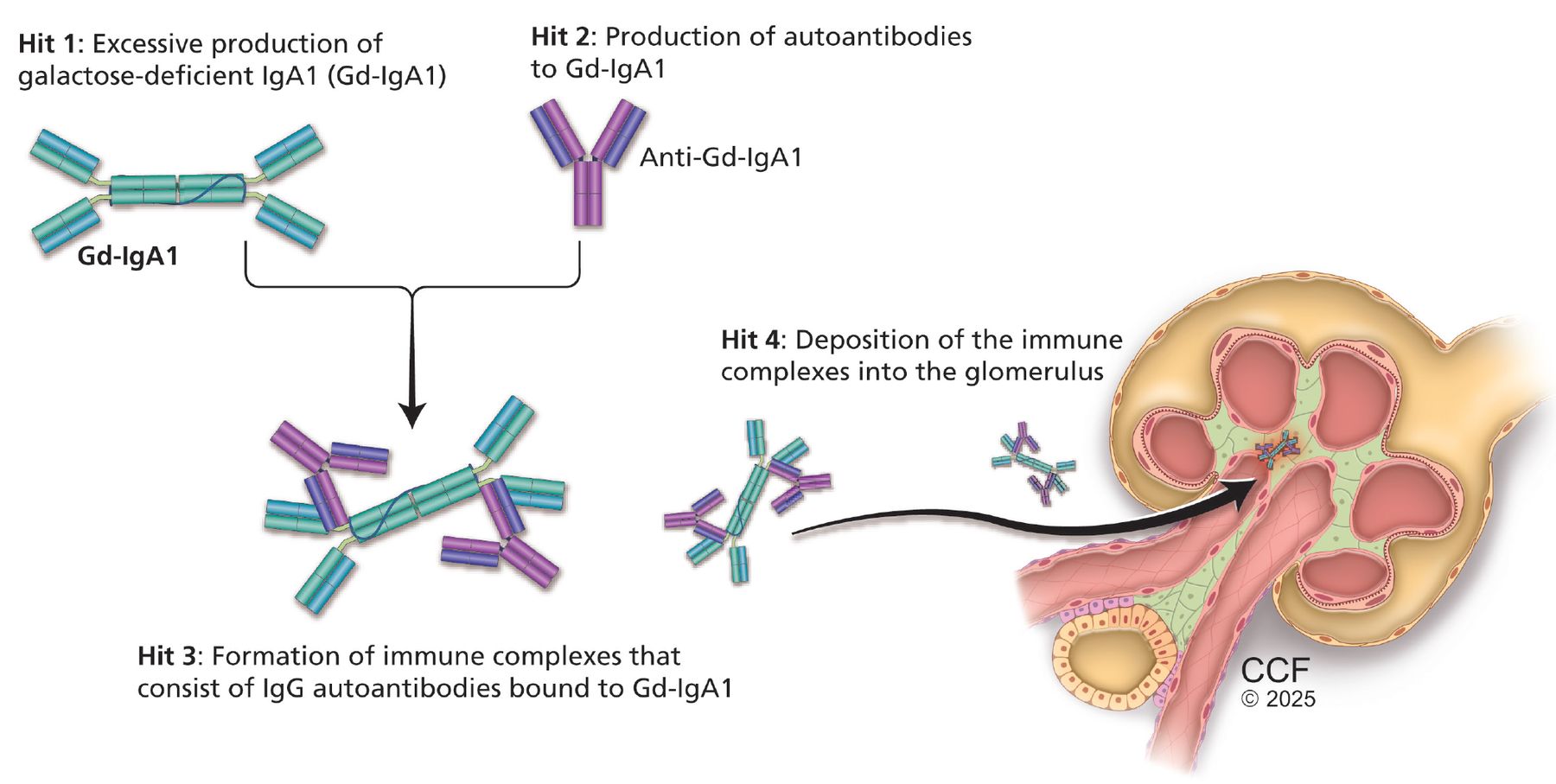
IgA nephropathy is an autoimmune disease of mucosal type IgA1 characterized by deposition of immune complexes in the glomerulus. Its pathogenesis is now firmly established and understood as the 4-hit hypothesis (Figure 1).1 The 4 hits comprise a complex interplay of genetic factors (involving polymorphisms in human leukocyte antigen, complement, and gut mucosal immunity) and environmental factors such as the gut microbiome, all of which contribute to the development of IgA nephropathy.

Pathogenesis of immunoglobulin (Ig) A nephropathy: the “4-hits” hypothesis.
Hit 1: excessive production of galactose-deficient IgA1
Galactose-deficient IgA1 in IgA nephropathy lacks the terminal galactose moieties at the hinge region of the molecule.1,4 The primary site for production of galactose-deficient IgA1 is now believed to be the gut and nasal mucosa.2 Many factors have been implicated in the production of galactose-deficient IgA1.
Genetics may influence the O-galactosylation of the IgA hinge region.5 Abnormal galactosylation of IgA can be an inherited trait, but this alone is insufficient for development of IgA nephropathy.
Cytokines, including serum B-cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL), are important regulators of mucosal B-cell survival and proliferation. BAFF and APRIL promote the formation of galactose-deficient IgA1–producing plasma cells in the mucosa.6
Alterations in the composition of the gut micro-biome (which communicates with mucosal-associated lymphoid tissue) have been implicated in IgA nephropathy.7 Mucosal dysbiosis may be related to dysregulated mucosal IgA synthesis. Recently, it was shown that patients with IgA nephropathy have a relative over growth of mucin-degrading bacteria,8 which are capable of deglycosylating IgA1.
Hit 2: production of autoantibodies to galactose-deficient-IgA1
Antibodies, either IgG or IgA, recognize the galactose-deficient hinge region of galactose-deficient IgA1, a neoepitope.1 Routine immunofluorescence on kidney biopsy detects IgA bound to galactose-deficient IgA1 as the predominant immune complex deposited; however, evidence supports the presence of IgG autoantibodies, which also play a role in the pathogenesis of the disease.
Hit 3: formation of immune complexes consisting of IgG autoantibodies bound to galactose-deficient IgA1
Clinical and histologic activity correlate with the level of circulating immune complexes.1,4 Additionally, alternative complement and terminal complement activity have been shown to correlate with the concentration of galactose-deficient IgA1.
Hit 4: deposition of immune complexes into the glomerulus
The effect of the immune complexes on mesangial cells within the glomerulus drives kidney injury.9,10 Deposition of immune complexes activates mesangial cells, leading to production of inflammatory molecules such as interleukin-6 and platelet-derived growth factor and complement, which signal infiltration of monocytes and mediate glomerular injury.
The role of complement
Complement plays a prominent role in mesangial injury and is a major driver of glomerular inflammation.11 C3 is deposited in the mesangium, activating both the alternative and lectin pathways. In vitro studies provide evidence of alternative pathway proteins such as complement components C5, C6, and C9 and other membrane attack complex antigens in the glomeruli of patients with IgA nephropathy, whereas markers for classical pathway activation such as C1q and C4 are less prominent. Complement factor H–related protein competes with the binding of factor H, a regulator protein, leading to an increase in the activity of the alternate complement pathway.
DIAGNOSIS REQUIRES CLINICAL SUSPICION AND KIDNEY BIOPSY
Despite advances in understanding the pathogenesis of IgA nephropathy, diagnosis requires a kidney biopsy. Clinical suspicion arises from the presence of acute kidney injury, hematuria, or proteinuria. Uncommonly, patients present with gross hematuria or synpharyngitic hematuria (hematuria with pharyngitis), a presentation seen more often in younger patients (< 40 years).1 In older populations, IgA nephropathy can be clinically occult with worsening kidney function and microscopic hematuria. While routine screening is common in countries with a high prevalence, such as Japan and China, there are no screening guidelines in the United States. Therefore, timely referral to nephrology upon discovery of hematuria or proteinuria is critical.
Histologic examination of the kidney biopsy specimen with immunofluorescence microscopy will show IgA deposits in the mesangium or capillary loops accompanied by mesangial changes (proliferation and expansion). Serologic markers, while extensively studied and now frequently used in clinical trials, require further validation before they can be applied in the clinic.
Alternative diagnoses must be considered when histopathology reveals IgA staining, as there are numerous mimics of primary IgA nephropathy (Table 1).1,12–14 Systemic disease states associated with IgA nephropathy, labeled secondary IgA nephropathy, include IgA vasculitis, viral infections (human immunodeficiency virus, hepatitis), autoimmune disease (inflammatory bowel disease, psoriasis), cirrhosis, IgA-dominant postinfectious glomerulonephritis, and proliferative glomerulonephritis with monoclonal IgA deposits.
Immunoglobulin (Ig) A nephropathy and its mimics
Once the diagnosis is established, the characteristic findings are used to determine prognosis and clinical outcomes. Secondary IgA nephropathy and IgA vasculitis have been largely excluded from clinical trials and carry a different prognosis than primary IgA nephropathy.
PROGNOSTIC TOOLS
Oxford Classification of IgA nephropathy
The Oxford Classification of IgA nephropathy was introduced in 2009.15 The purpose was to create a standardized histopathologic scoring system using 4 variables that correlate most strongly with patient outcomes, in addition to showing adequate agreement among nephropathologists. The variables are mesangial hypercellularity (M), endocapillary hypercellularity (E), segmental glomerulosclerosis (S), and tubular atrophy/interstitial fibrosis (T), reported as the MEST score. The system was updated in 2016 to incorporate crescents (C) to further aid in predicting renal outcomes (Table 2).15,16
Oxford Classification of immunoglobulin A nephropathy: MEST-C score
M, S, and T were found to be independent predictors of glomerular filtration rate (GFR) decline in the original Oxford cohort, but E lesions were not conclusively predictive of decline.17 Similar associations in GFR decline were seen in patients with endocapillary hypercellularity (E) independent of immuno-suppression.18,19 The Oxford cohort did not control for immunosuppression, leading to a treatment bias. Further, patients with E lesions were more likely to receive immunosuppression. Collectively, this evidence supports the perception that endocapillary lesions are responsive to immunosuppressive treatment and contribute to the decline of kidney function if not treated with immunosuppression.
Although helpful for diagnostic standardization and prognosis, this scoring system does not consider the presence of hypertension, degree of proteinuria, or reduced GFR.
International IgA nephropathy risk prediction tool
The introduction of the international IgA nephropathy risk prediction tool further refines risk stratification by integrating histologic and clinical factors to predict renal outcomes at the time of biopsy and up to 7 years.20 It was derived in a multiethnic international cohort with biopsy-proven idiopathic IgA nephropathy and is designed to predict the risk of a 50% decline in estimated GFR or end-stage kidney disease after biopsy.
This web-based prediction tool includes the estimated GFR at the time of biopsy, systolic and diastolic blood pressure at the time of biopsy, proteinuria, age, race, use of angiotensin-converting enzyme inhibitors or angiotensin receptor blockers, MEST score, immunosuppression use at or before kidney biopsy, and the number of months after a kidney biopsy that the clinician will determine the risk of progressive IgA nephropathy.
Criticisms of the IgA nephropathy prediction tool include its lack of dynamic longitudinal monitoring ability and the absence of modern therapies (endothelin receptor antagonists and sodium-glucose cotransporter [SGLT] 2 inhibitors). Also, it was not validated to guide the use of immunosuppression.20
Proteinuria as an indicator of kidney function
The goal of therapy in IgA nephropathy, as in all kidney disease, is to prevent progression to end-stage kidney disease by decreasing the rate of GFR loss. The main therapeutic targets in IgA nephropathy include reducing proteinuria and controlling blood pressure. The severity of proteinuria remains the strongest indicator of kidney outcome. The KDIGO (Kidney Disease: Improving Global Outcomes) guidelines21 recommend reducing proteinuria to less than 1 g per day as a surrogate marker for improved kidney outcome, and consideration of immunosuppressive therapy if unable to achieve proteinuria levels lower than 1 g per day with conservative management such as RAAS blockade. However, recent large registry data have revealed that 30% of patients with time-averaged proteinuria of 0.44 to less than 0.88 g/g (of creatine) developed kidney failure within 10 years.22 It is therefore clear that patients with IgA nephropathy and lower degrees of proteinuria may benefit from more intensive disease management.
Currently, the goal of therapy is proteinuria of less than 0.5 g and absence of hematuria. These targets have not been well studied in a prospective therapeutic trial, however, and we do not yet know the risk or benefit of attempting to achieve such targets. Several new trials and therapeutics have emerged that require an update to our approach to diagnosis and treatment of IgA nephropathy.
CURRENT TREATMENT OPTIONS
Nonimmunosuppressive therapy
Treatments targeting RAAS reduce proteinuria and preserve nephrons across the spectrum of glomerular diseases, including IgA nephropathy. Use of angiotensin-converting enzyme inhibitors or angiotensin receptor blockers carries a strong recommendation in the most recent KDIGO guidelines,21 along with a target blood pressure of 120/70 mm Hg or lower and lifestyle modifications that include smoking cessation, weight reduction, salt restriction (< 2 g/day), and exercise. KDIGO guidelines no longer recommend fish oil for IgA nephropathy.
New nonimmunosuppressive therapy options include the SGLT-2 inhibitors and dual endothelin receptor and angiotensin receptor antagonists.
SGLT-2 inhibitors. There have been no dedicated trials to evaluate IgA nephropathy outcomes with the use of these agents. However, IgA nephropathy was well represented in DAPA-CKD (Dapagliflozin in Patients With Chronic Kidney Disease),23 a randomized controlled trial that evaluated the effect of dapagliflozin in patients with chronic kidney disease and albuminuria due to various causes. In a prespecified analysis of DAPA-CKD, 270 patients with IgA nephropathy treated with dapagliflozin had a 26% reduction in proteinuria compared with placebo. Additionally, the primary outcome (sustained decline in estimated GFR of 50% or more, end-stage kidney disease, or death from a kidney disease–related or cardiovascular cause) occurred in only 6 (4%) participants on dapagliflozin vs 20 (15%) on placebo (hazard ratio 0.29; 95% confidence interval [CI] 0.12–0.73), offering a 71% risk reduction. Criticisms of this trial include the lack of adequate blood pressure control with angiotensin-converting enzyme inhibitors or angiotensin receptor blockers in the run-in period compared with other trials in IgA nephropathy, in addition to recruitment of older patients and exclusion of patients with recent immunosuppression use.
The safety and efficacy of the dual endothelin and angiotensin receptor antagonist sparsentan in IgA nephropathy was recently evaluated in the PROTECT (Efficacy and Safety of Sparsentan Versus Irbesartan in Patients With IgA Nephropathy) trial.24 In this large randomized, active-controlled study, adults with high-risk IgA nephropathy (> 1 g proteinuria per day) received sparsentan or irbesartan 300 mg daily. The primary efficacy end point was a change from baseline to week 36 in the urine protein-creatinine ratio based on a 24-hour urine sample. The sparsentan group saw a 49.8% proteinuria reduction compared with 15.1% in the irbesartan group, which was maintained until the 110-week trial ended. At 2 years, the estimated GFR chronic rate of change (from weeks 6 to 110) was −2.7 mL/min/1.73 m2/year with sparsentan and −3.8 mL/min/1.73 m2/year with irbesartan (difference 1.1 mL/min/1.73 m2/year, 95% CI 0.1–2.1).
The rate of adverse events was similar in the 2 groups, with more hypotension and acute kidney injury occurring in the sparsentan group. Due to the potential hepatotoxicity and fetal toxicity of endothelin receptor antagonists, the FDA requires the Risk Evaluation and Mitigation Strategy for sparsentan, mandating liver function monitoring for patients on the drug and, for those capable of becoming pregnant, maintaining contraception while on treatment and 1 month after. RAAS blockers should be stopped when converting to sparsentan.
On the strength of the 36-week data showing proteinuria reduction, the FDA granted accelerated approval to sparsentan for patients with IgA nephropathy deemed high risk for progression; recently, the drug obtained full approval.
It is currently not known whether the addition of SGLT-2 inhibitors to dual endothelin receptor and angiotensin receptor antagonists or endothelin receptor antagonists will add further proteinuria reduction and estimated GFR benefit. The results of an open-label extension of the PROTECT trial are awaited.
The endothelin receptor antagonist atrasentan was recently granted accelerated approval based on findings from the phase 3 ALIGN (Atrasentan in Patients With IgA Nephropathy) trial.25
Immunosuppressive therapy
Systemic corticosteroids are frequently used in IgA nephropathy, yet their role in management of this disease is controversial. Several randomized controlled trials and meta-analyses that examined corticosteroid use in IgA nephropathy have had conflicting results. Modern randomized controlled trials such as STOP-IgAN (Supportive Versus Immunosuppressive Therapy for the Treatment of Progressive IgA Nephropathy)26 and TESTING (Therapeutic Evaluation of Steroids in IgA Nephropathy Global)27 have best represented the use of systemic corticosteroids and their risks, which were likely underreported in older studies.28
STOP-IgAN26 was a relatively small randomized controlled trial testing the safety and efficacy of immunosuppressive therapy combined with supportive care compared with supportive care alone. Immunosuppressive therapy consisted of corticosteroids for those with estimated GFR of 60 mL/min/1.73 m2 or greater, and cyclophosphamide followed by azathioprine and corticosteroids for those with estimated GFR between 30 and 59 mL/min/1.73 m2. Of the 337 patients entering the run-in phase, 106 responded to supportive care after 6 months, which included RAAS blockade, smoking cessation, and cholesterol-lowering with statins; these were not randomized. Only 5% in the supportive-care arm reported complete remission (ie, urine protein-creatinine ratio < 0.2 g/24 hours and stable renal function with a fall in estimated GFR < 5 mL/min/1.73 m2 from baseline) compared with 17% in the immunosuppressive arm. At the end of the 3-year trial, there was no difference in estimated GFR between the groups. Not surprisingly, immunosuppression with corticosteroids saw higher rates of weight gain, impaired glucose tolerance, and serious adverse events such as infection. STOP-IgAN therefore solidified the value of supportive or nonimmunosuppressive care in IgA nephropathy.
The TESTING trial was a randomized clinical trial comparing oral methylprednisolone (0.6–0.8 mg/kg/day for 2 months and then tapering, with a treatment period of 6 to 8 months) with placebo, carried out in a predominantly East Asian population.27 While methylprednisolone resulted in a lower likelihood of the primary end point (40% decline in estimated GFR, end-stage kidney disease, or death due to kidney failure), it came at the price of serious infections, including 2 infection-related deaths, and the investigators suspended the trial.
The trial resumed recruitment after the methyl-prednisolone dose was reduced (0.4 mg/kg/day for 2 months, tapered over 6 to 9 months) and prophylactic antibiotics were mandated.29 The primary composite end point occurred in 28.8% (74 patients) of the methylprednisolone group vs 43.1% (106) of the placebo group (hazard ratio 0.53, 95% CI 0.39–0.72, P < .001) over a mean follow-up of 4.2 years. Despite the reduced steroid dose, serious adverse events were 4 times higher in the methylprednisolone group than in the placebo group: 37 vs 8 total events that occurred in 28 (10.9%) vs 7 (2.8%) participants.
Finally, targeted-release formulation (TRF) budesonide is the only immunosuppressive drug fully approved by the FDA to treat IgA nephropathy. The hypothesis is that TRF budesonide is delivered directly to the small bowel and Peyer patches, where the galactose-deficient IgA is produced, interrupting a key mediator of IgA nephropathy. In theory, because of extensive first-pass metabolism, less drug would reach the systemic circulation and limit glucocorticoid toxicity.
The NefIgArd (Efficacy and Safety of Nefecon in Patients With Primary IgA [Immunoglobulin A] Nephropathy) trial,30 a phase 3 randomized trial, evaluated TRF budesonide vs placebo in patients with proteinuria of 1 g or more over a 9-month period. TRF budesonide resulted in significantly reduced proteinuria and sustained estimated GFR benefit over a 2-year follow-up. However, like other trials of systemic corticosteroids for IgA nephropathy, the proteinuria returned after TRF budesonide was stopped, and steroid-related side effects were more common in the TRF budesonide group, including weight gain, facial edema, acne, peripheral edema, and hypertension.
Treatment recommendations
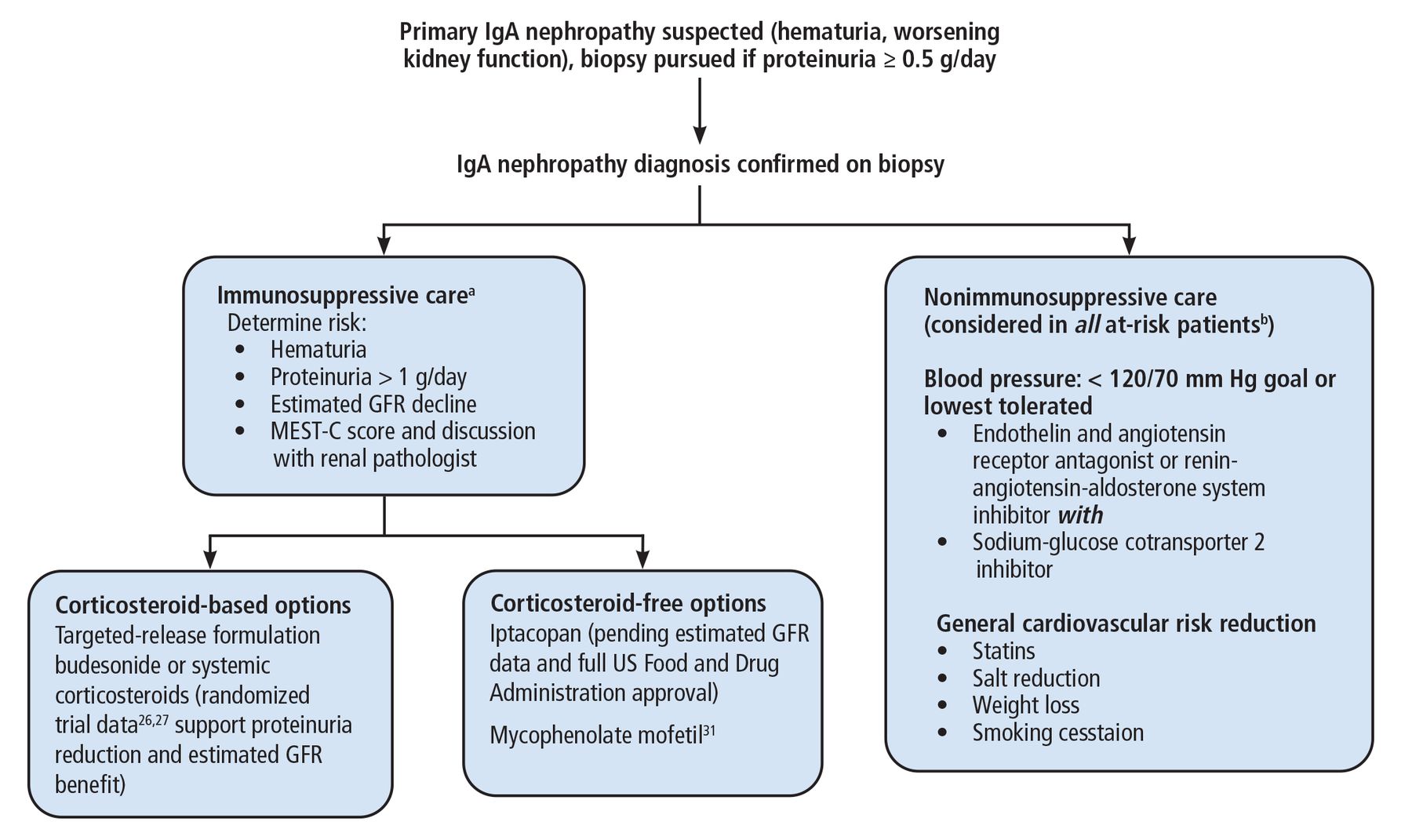
The landscape for treatment of IgA nephropathy has changed rapidly and will continue to change in the coming months and years. Figure 226,27,31 presents the authors’ recommended approach, with these considerations:
A proteinuria threshold of 0.5 g/day is the new cutoff for warranting a biopsy, as opposed to the traditional value of 1 g/day or greater.
While the MEST-C score cannot be used to guide immunosuppressive therapy, we advise considering it in addition to a direct discussion with the renal pathologist who interpreted the biopsy.
Nonimmunosuppressive therapy should be considered in conjunction with immunosuppressive therapy.
There is inadequate evidence to support superiority of TRF budesonide over systemic corticosteroids, and this decision is made on a case-by-case basis.

Our approach to immunoglobulin (Ig) A nephropathy.
aWe monitor patients receiving immunosuppressive therapy with assessment of blood pressure and protein-creatinine ratio, renal function panel, and urinalysis every 3 months.
bThose with proteinuria > 0.5 g/day.
GFR = glomerular filtration rate; MEST-C = mesangial hypercellularity, endocapillary proliferation, segmental glomerulosclerosis, tubulointerstitial fibrosis, crescents
Note that this approach can include use of mycophenolate mofetil as a corticosteroid-free immunosuppressive option. A randomized trial of 170 Chinese patients with IgA nephropathy showed mycophenolate mofetil when added to supportive care (renin-angiotensin system blockade) reduced the risk of the primary composite outcome (doubling of serum creatinine, end-stage kidney disease, or death due to kidney or cardiovascular cause) compared with supportive care alone.31
FUTURE TREATMENT OPTIONS
A variety of treatment options are under investigation for the management of IgA nephropathy targeting the different “hits” in the pathogenesis model (Table 3).24,25,30,32–47
Recently approved and future treatment options for immunoglobulin A nephropathy
Complement inhibitors. Significant research is focused on the complement cascade, reflecting the key role of the complement system in the development of IgA nephropathy. Several complement inhibitors are being studied in phase 2 and 3 trials, with mixed results.32–40 There has been much focus on inhibition of the alternative pathway of complement, which impacts the deposition of immune complexes in the glomerulus (the fourth hit in the pathogenesis model of IgA nephropathy).
Iptacopan (LNP023), an oral factor B inhibitor that prevents the activity of the alternative pathway C3 convertase, was evaluated in 66 patients with IgA nephropathy in a phase 2 trial.32 At 6 months, participants who received iptacopan 200 mg twice daily had a 40% reduction in proteinuria compared with placebo. In a follow-up phase 3 trial, iptacopan showed a significant reduction in proteinuria at 9 months compared with placebo.38 It was recently granted accelerated approval by the FDA.
The complement inhibitor class of drugs will likely be used in cases of IgA nephropathy that are resistant to traditional treatments, including corticosteroids, and have a significant inflammatory component on kidney biopsy, or those where a steroid-sparing regimen is ideal. There is interest in correlating the intensity of C3 staining on immunofluorescence microscopy of kidney biopsies and the potential response to complement inhibition.
Inhibition of antibody-producing B cells (targeting the second and third hits in IgA nephropathy pathogenesis) has also emerged as a therapeutic target for the management of IgA nephropathy. While rituximab has not been shown to be beneficial, other B-cell receptor targets have shown some initial success, including APRIL (a proliferation-inducing ligand), BAFF (B-cell activating factor), and plasma cell receptors.48,49 APRIL and BAFF regulate B-cell survival.
APRIL may help to specifically produce IgA1 molecules by controlling the immunoglobulin class switch recombination.48,49 Several monoclonal antibodies against APRIL are currently under investigation, including sibeprenlimab, zigakibart, and atacicept.41–45 A phase 2b clinical trial of atacicept showed a significant reduction in proteinuria compared with placebo, and a phase 3 clinical trial is under way.44
Antiplasma cell therapies (second and third hits in IgA nephropathy pathogenesis) are also being investigated as potential treatment options.46,47 Monoclonal antibodies to CD38 (felzartamab and mezagitamab) are being assessed in early-stage clinical trials.46 Larger studies are needed to assess the efficacy of this approach in the management of IgA nephropathy.
NEW UNDERSTANDING AND NEW CHALLENGES
Advances in our understanding of the pathogenesis, prognosis, and, most important, therapeutic options for IgA nephropathy have been significant. For decades, treatment options have been limited, with many reaching end-stage kidney disease within their lifetime. Recognition of the disease still depends on urinalysis and quantification of proteinuria, but with new therapies on the horizon, there is hope that awareness will increase.
For the treating clinician, the management of IgA nephropathy is a complex clinical scenario. The 4-hit model provides a blueprint for the pathogenesis, allowing targeted management of the disease, but appropriate use of novel therapies and assessment of response remain significant challenges. Among the questions to consider are the following:
How should these drugs be combined, if at all?
How long should each therapy be given?
Do newer therapies result in a true reduction in the rate of end-stage kidney disease?
Also important are conversations for patients and clinicians on cost and access to therapy. Ongoing study, debate, and conversation within the nephrology community are needed to prioritize these novel therapies and develop guidelines.
DISCLOSURES
Dr. Cohen has disclosed consulting for Gilead Sciences, Inc. Dr. Cavanaugh has disclosed consulting for Cerium Pharmaceuticals, Travere Therapeutics, and Vera Therapeutics, and serving as an advisor or review panel participant for Cerium Pharmaceuticals. Dr. Ramsawak and Dr. Linares report no relevant financial relationships which, in the context of their contributions, could be perceived as a potential conflict of interest.
- Copyright © 2025 The Cleveland Clinic Foundation. All Rights Reserved.
REFERENCES
In this issue




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.