


I am flattered to present the inaugural talk in the biostatistics and clinical research design series on the architecture of clinical research. This content is based on the teachings of my mentor, Dr. Alvan Feinstein, who together with Dr. David Sackett, is credited with pioneering clinical epidemiology. Dr. Feinstein was a Sterling Professor at the Yale School of Medicine. His main opus of work is a book called, Clinical Epidemiology: The Architecture of Clinical Research.1 This paper is named in credit to Dr. Feinstein’s enormous contribution. I will review some important terms defined by Dr. Feinstein to provide the background necessary for the remainder of the talks in this series.
To start, I will frame this topic by asking the following question: Why do we do research? I’ll talk about the basic structure of research studies and provide a taxonomy, as Dr. Feinstein would say, a nomenclature with which to understand trial design and the sources of bias in those trials. Then, I will discuss these sources of bias in detail using the taxonomy that Dr. Feinstein described in his aforementioned book. Finally, I will share with you some examples of bias in clinical trials to help you better understand these concepts.
Now, the answer to the basic question posed above is: basically, we do cause-and-effect research to establish the causality of a risk factor or the efficacy of a therapy. Does cigarette smoking cause lung cancer? Does taking hydrochlorothiazide help systemic hypertension? Does air pollution worsen asthma? Does supplemental oxygen help patients with chronic obstructive pulmonary disease (COPD)?
Cause-and-effect research can be subsumed under 2 broad issues: causal risk factors and therapeutic efficacy. In his review of early false understandings in medicine that were based on anecdotal observation alone, Thomas cites many examples—“the undue longevity of useless and even harmful drugs can be laid at the door of authority,” ie, empiricism, lack of rigorous research.2 The field is full of these: yellow fever causality, the value of cupping, and even intermittent mandatory ventilation when it was described by John Downs in 1973 and touted as a superior mode for weaning patients from mechanical ventilation.3 Twenty-five years later, randomized controlled trials by Brochard et al4 indicated not only that intermittent mandatory ventilation was not the best mode to wean but was, in fact, the worst mode for weaning patients from mechanical ventilation compared with either pressure support or spontaneous breathing trials. Many more examples exist to demonstrate the false understandings that can be ascribed to lack of rigorous study or evidence in medicine.
Before systematically exploring the sources of bias in Feinstein’s construct, let us define some very basic terms from his book. Dr. Feinstein talks about the baseline state, which refers to the group of patients under study who are culled from a larger population to whom the results are intended to be applied (Figure 1).1 This baseline group is hopefully representative of this larger target population. As a nod to the later discussion, Dr. Feinstein would call bias introduced by unusual assembly of the study population from the larger intended population as “assembly bias.” So, if the group under study is not representative of either the patients you see or the world of patients with this condition or if there is something special or distinctively nonrepresentative about the study population, then the results may be subject to “assembly bias.” Assembly bias can compromise the so-called “external” validity of the study—its ability to be applied to populations beyond the study group.
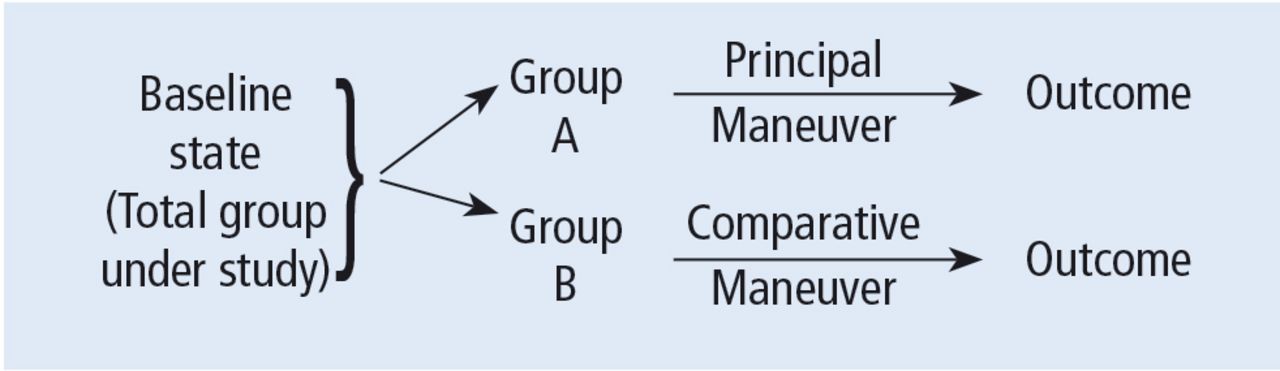
Design of a controlled trial according to Feinstein.1
Having assembled a baseline group for study, that group is classically allocated to 1 of 2 (or sometimes more than 2) compared therapies. In a controlled trial, patients can be allocated using a variety of strategies, including randomization. Using the paradigm diagram (Figure 1, which considers a 2-arm trial), patients are allocated to 1 of 2 compared groups—group A and group B. Then, in a treatment trial, 1 group receives the principal maneuver, which is the drug or intervention under study—for example, supplemental oxygen for patients with COPD. The comparative maneuver is allocated to group B, which also receives all the other treatments (called “co-maneuvers”) that are used to treat the condition under study. In a trial of supplemental oxygen for COPD evaluating lung function and exacerbation frequency as outcome measures, such co-maneuvers might include inhaled bronchodilators, inhaled corticosteroids, pulmonary rehabilitation, and Pneumovax vaccine. Ideally, these co-maneuvers are equally distributed between the compared groups (A and B).
So, in summary, we have a comparative maneuver, which is the nonadministration of supplemental oxygen in this proposed trial of supplemental oxygen in COPD, the principal maneuver—administration of oxygen—and all the co-maneuvers that are ideally equally distributed between both groups. This balanced distribution of co-maneuvers between the compared groups helps to ensure that any differences in the study outcome measures (ie, what is counted as the main impact of the intervention under study) can be solely attributed to the principal maneuver. When this condition—that the difference in outcomes can be reliably ascribed to the study intervention—is satisfied, the study is felt to be “internally” valid. As we will see, ensuring internal validity requires freedom from the many sources of what Dr. Feinstein calls “internal bias.”
Back to basic terms: “cohort” in Dr. Feinstein’s language is a group that shares common traits and is followed forward in a longitudinal study. The “outcome measure” is self-evident—it is what is being measured, with the “primary outcome” being the predefined measure that is considered the most important (and ideally most clinically relevant) impact of the study intervention. Later in this series of lectures, there will be discussions of power calculations and the so-called “effect size”—the magnitude of effect that the intervention is expected to produce and that is ideally deemed clinically important.
An important consideration in designing a trial is to define and declare the primary outcome measure carefully because defining the primary outcome measure has important implications for the study. I will provide an example from the alpha-1 antitrypsin deficiency literature. Some of you have probably read what has been called the RAPID trial.5 RAPID was a trial of augmentation therapy vs placebo in patients with severe alpha-1 antitrypsin deficiency. The primary outcome measure (which was pre-negotiated with the US Food and Drug Administration [FDA]) was computer tomography (CT) lung density determined at functional residual capacity (FRC) and total lung capacity (TLC). The trial failed to achieve statistical significance in regard to CT lung density, although the study authors argued that CT density measurements made at TLC were more reproducible than those made at FRC. When the results were analyzed by TLC alone, the results were statistically significant, but when they were analyzed with FRC and TLC combined, they were not. In the end, based on the pre-negotiated primary outcome measure of CT density based on both FRC and TLC, the FDA rejected the proposal for a label change to say that augmentation therapy slowed the loss of lung density even though the weight of evidence was clearly in its favor. This case exemplifies just how critical the choice of primary outcome measure can be.
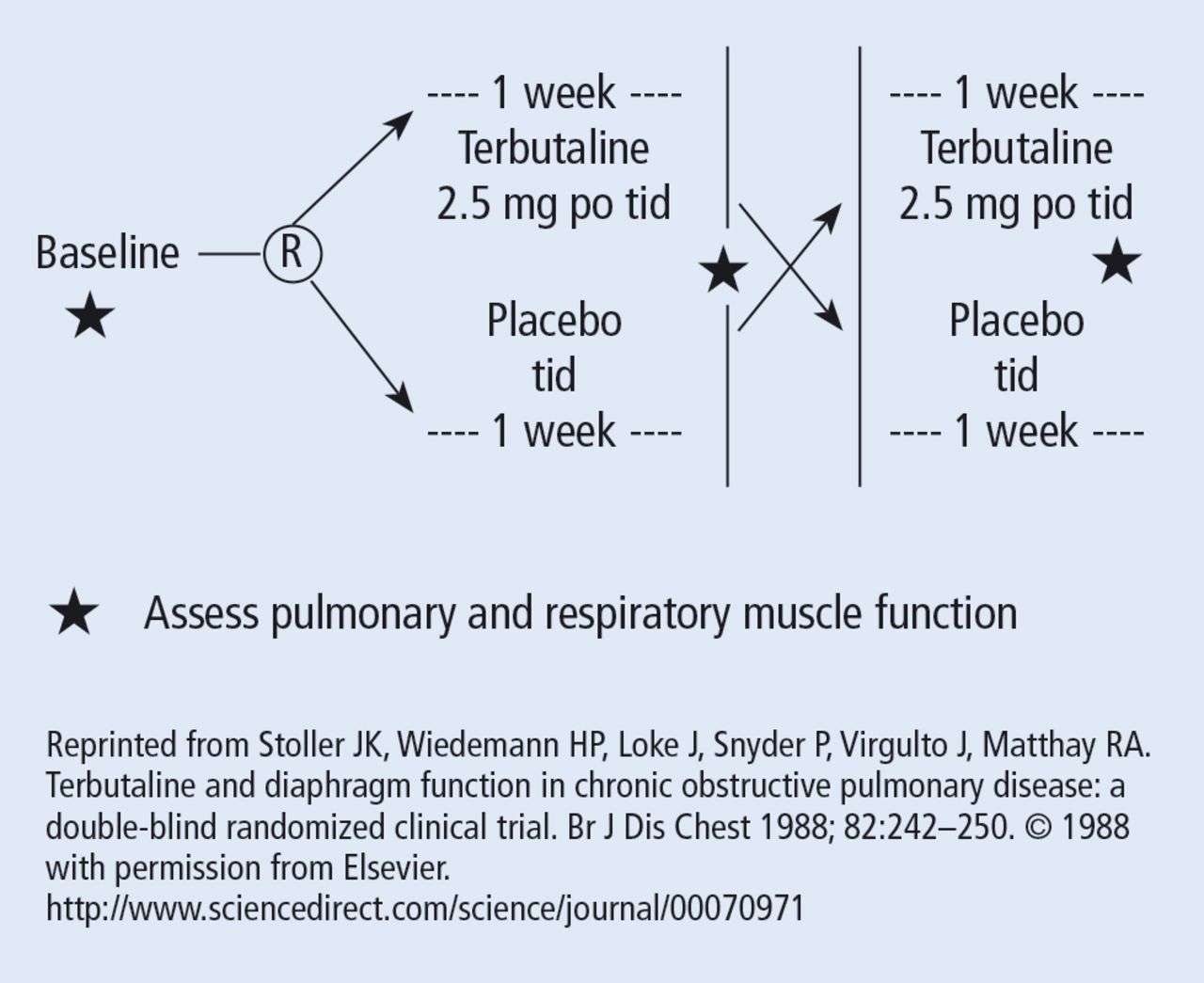
The wash-out period refers to an interval in a subset of randomized trials called “crossover trials” in which the primary intervention is discontinued and the patient returns to his baseline state before the comparative maneuver is then implemented (Figure 2).6 In order to perform a crossover trial, it is important that the effects of the initial intervention can “wash out” or be fully extinguished. So, for example, in trials of radiation therapy vs surgery, it is impossible to do a crossover trial because the effects of radiation can never completely wash out nor can those of surgery, which are similarly permanent. For example, we cannot replace the colon once it is resected for cancer or replace the appendix once removed. Therefore, producing a wash-out requires some very specific pharmacokinetic and pharmacodynamic features in order for a crossover trial to be considered. Later talks in this series will discuss the enhanced statistical power of a crossover trial, where one is comparing every patient to him or herself rather than to another patient.
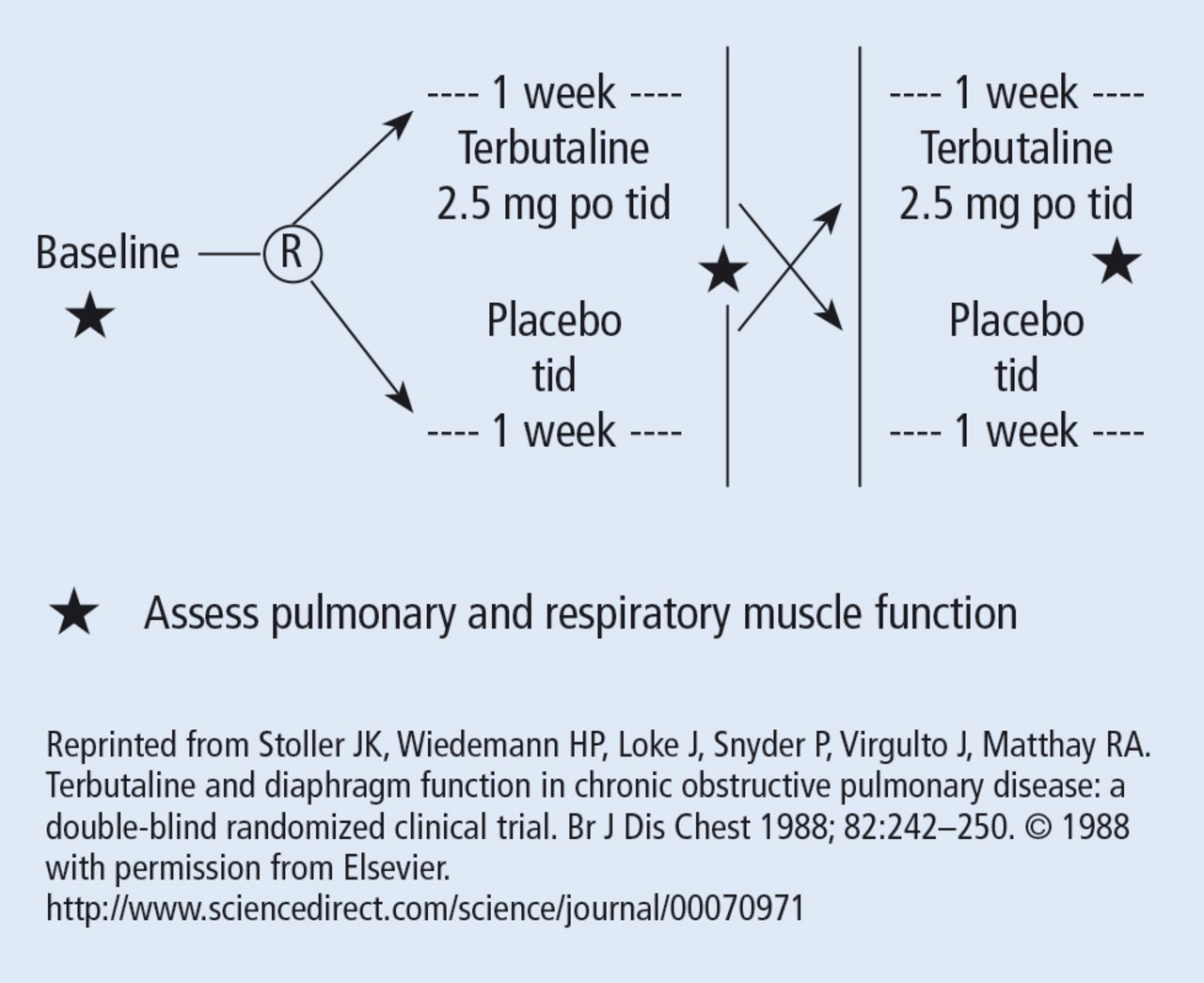
Design of a randomized crossover trial of terbutaline for diaphragmatic function. The wash-out period separates the first and the second interventions (begins at the star in the diagram).
So, there is always an appetite to do a crossover trial as long as the criteria for wash-out can be met, namely again that the primary intervention can dissipate completely to the baseline state before the alternative intervention is implemented.
“Placebo” is a fairly self-evident and well-understood term; placebo refers to the administration of a maneuver in a way that is identical to the principal maneuver except that the placebo is not expected to exert any clinical effect.
“Blinding” is the unawareness of either the investigator or of the patient to which the intervention is being administered. “Single-blinding” refers to the condition in which either the study or the investigator (but not both) is unaware, and “double-blinding” refers to the condition in which both the subjects and the investigators are unaware. There can be some subtle issues that compromise whether the patient is aware of the intervention that he or she is receiving and that can potentially condition the patient’s response, particularly if there is any subjective component of the assessment of the outcome. So, blinding is important.
With these terms describing the elements of a clinical study now described, let us turn to the types of studies that comprise clinical research. The first group of study types is what Dr. Feinstein called descriptive studies—studies that simply describe phenomena without comparison to a control group. As an example of a descriptive study, Sehgal et al7 recently described the workup of a focal, segmental pneumonia in a patient taking pembrolizumab for lung cancer. In this paper, there were four other cases of focal pneumonia accompanying pembrolizumab use that were assembled from the literature, making this descriptive paper a so-called case series. A “case series” differs from a “single case report,” which reports a single patient experience. Though limited in their ability to establish cause and effect, case reports and case series can help researchers develop proof of principle, so I would not discount the value of case reports.8
I can cite a case report from of my own experience that demonstrates this point. In 1987, I saw a patient from Buffalo who had primary biliary cirrhosis and the hepatopulmonary syndrome (HPS). She was so debilitated by her HPS that she could not stand up without desaturating severely. Although she had normal liver synthetic function, she was severely debilitated by her HPS and the decision was made to offer her a liver transplant, which, at that time, was considered to be relatively contraindicated. Much to everyone’s amazement and satisfaction, her HPS completely resolved after the transplant surgery. Her oxygenation and alveolar-arterial oxygen gradient normalized, and her clubbing resolved. We reported this in a case report, which began to affect the way people thought about the feasibility of liver transplant for the HPS.8 The lesson is: do not underestimate the power of a thoughtful case report.
The second group of research study types is called “cohort studies,” in which one actually compares outcomes between 2 groups in the study. Cohort studies fall into the bucket of either “observational cohort studies,” in which allocation to the compared maneuvers is not performed by randomization but by any other strategy, and “randomized trials.” In observational studies, allocation could occur through physician choice, as when the physician prescribes a treatment to 1 group but not another, or by patient choice or circumstance. For example, an observational cohort study of the risk of cigarette smoking would compare outcomes between smokers and non-smokers where the patient choses to smoke under his/her own volition. Alternatively, the circumstances of an exposure could allocate someone to the principal maneuver, as when we are studying the effect of exposure to World Trade Center dust in the firefighters who responded or of exposure to nuclear radiation in Hiroshima survivors. These are examples of observational cohort studies that compare exposed individuals to unexposed individuals, where the exposure did not occur by randomization but by choice or unfortunate circumstance.
In contrast to observational studies, allocation in randomized trials occurs through a formal process. Randomization has the specific purpose of attempting to ensure that patients are allocated to 2 comparative groups from the baseline group with comparable risk for developing the outcome measure. When randomization is effective, differences in study outcomes can be reliably ascribed to the intervention rather than to differences in the baseline susceptibility of the compared groups.
While randomization is an excellent strategy to ensure baseline similarity between compared groups, randomization can fail, and its effectiveness must be checked. Specifically, in a randomized trial, it is customary to examine the compared groups at baseline on all features that can affect the likelihood of developing the outcome measure. If the groups turn out to be dissimilar at baseline in an important way, then the study is at risk for bias, which is specifically called “susceptibility bias” in Feinstein’s construct. Obviously, the larger number of baseline clinical and demographic features that can condition the likelihood of developing the outcome measure, the more difficult it is to achieve baseline similarity between compared groups and the more important it becomes to ensure that randomization has been effective. In this circumstance, larger numbers of participants in both compared groups are generally needed. More about susceptibility bias later.
There are generally 2 types of randomized trials: the so-called “parallel controlled trials” in which each group receives either the principal or the comparative maneuver and is followed and “crossover trials” in which each compared group receives both the principal maneuver and the co-maneuver at different times after an effective wash-out period. Wash-out was discussed above. Figure 2 shows an example of a crossover trial examining the effects of terbutaline on diaphragmatic function.6 The investigators administered terbutaline for a week, measured transdiaphragmatic pressures, gave the patient a terbutaline vacation (the “wash-out period”), and then crossed over those patients who were initially receiving terbutaline to placebo and initial placebo recipients to terbutaline, having remeasured diaphragmatic function after the wash-out period to assure that the patient’s diaphragmatic function prior to the second crossover was identical to his/her baseline state. If this return to baseline is accomplished, then the criteria from effective wash-out are satisfied.
Now, with these basic structural terms of clinical research defined, bias will occupy the remainder of the discussion. By definition, bias in a clinical trial is any factor in the design or conduct of the trial, either external to the trial or internal to the trial, that can alter the results in a way that either threatens the reliability of attributing the differences in outcomes between the compared groups with the principal maneuver (“internal validity”) or limits the ability of the results, however internally valid, to be applied to a specific population beyond the study group (“external validity”) (Table 1).1 This again is because the main goal of cause-and-effect research is to make sure that you can attribute differences between the 2 compared groups at the end of the trial to the intervention under study and nothing else.
Types of bias in a clinical trial according to Feinstein1
As we begin to talk about sources of bias, consider a study in which we compare survival of patients allocated to surgery vs nonsurgical therapy for lung cancer (Figure 3).1 This study is subject to the first type of so-called “internal bias” in the Feinsteinian construct—so-called “selection bias.” For example, all patients treated surgically were considered healthy enough by their doctors to undergo surgery, whereas patients treated without surgery may have been deemed inoperable because of comorbidities, lung dysfunction, cardiac dysfunction, and so on. If the results of such a comparison show that the mortality rate among surgical patients in this study was lower, the question then becomes: is the improved survival in surgical candidates due to the superior efficacy of surgery vs other therapy or was the enhanced survival due to the surgical patients being healthier to begin with? You can intuitively sense that the answer to this question is that the enhanced survival may be due to the better health of patients treated surgically rather than to the surgery itself because of how the patients were selected to receive it. So, this is a simple example of what Dr. Feinstein would call “susceptibility bias.” Susceptibility bias occurs when the 2 baseline groups are not comparably at risk or susceptible to developing the outcome measure, leading the naïve investigator in this specific example to attribute the difference in outcomes to the superiority of surgery when in fact it may have nothing to do with the surgery vs. the other maneuver. When susceptibility bias is in play, the difference between the outcomes in the compared groups could be attributed to the baseline imbalance of the groups rather than to the principal maneuver itself.

A comparison of surgery vs nonsurgical therapy for advanced lung cancer. An example of possible susceptibility bias.1
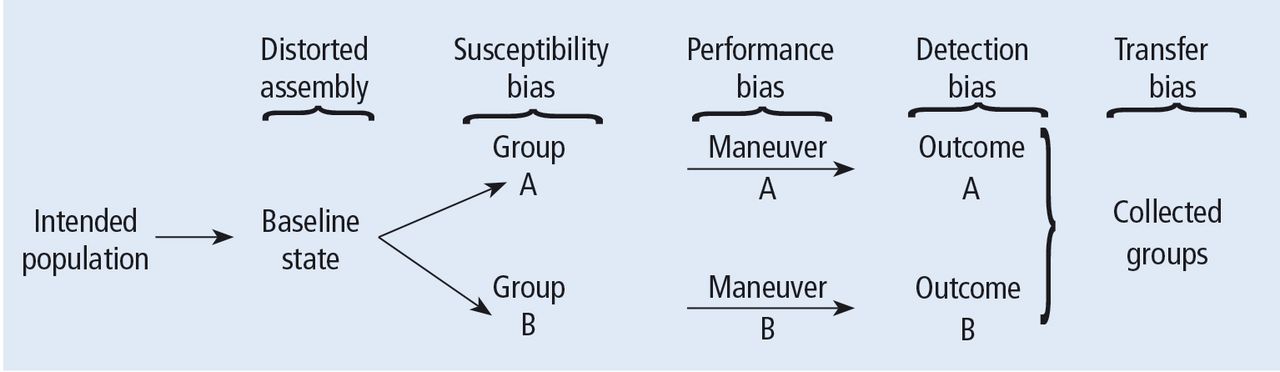
Turning back to the taxonomy of bias, there are four types that can threaten internal validity—“susceptibility,” “performance,” “detection,” and “transfer” bias—and 1 type of bias (called “external bias”) that can affect the generalizability of the study called “assembly bias” (Table 1).
Figure 4 shows where these various sources of bias appear in the architecture of a clinical trial. As just discussed, susceptibility bias affects the baseline state and the comparability of the groups. Performance bias relates to how effective and how comparably the co-maneuvers are given and whether the primary intervention is potent enough to affect an outcome. Both transfer and detection bias operate in detecting the outcome, especially regarding the rigor and frequency with which they are investigated. Transfer bias has to do with selective loss to follow-up of those included in the trial. If there is a systematic reason for loss to follow-up that is related to the impact of the intervention, then the study is at risk for transfer bias. For example, in a randomized trial of drug A vs placebo for pneumonia, if drug A is effective but all the drug A recipients fail to follow-up because they feel too good to return for follow-up, then transfer bias could be causing the study to show nonefficacy even though the drug works. So, if those who respond favorably are systematically lost to follow-up, and if all the patients who felt lousy wanted to see the doctor and came back for follow-up, such transfer bias would bias towards nonefficacy. Specifically, only patients remaining in the trial would be those who failed to respond and that would dilute any difference between the 2 groups despite the active efficacy of drug A.
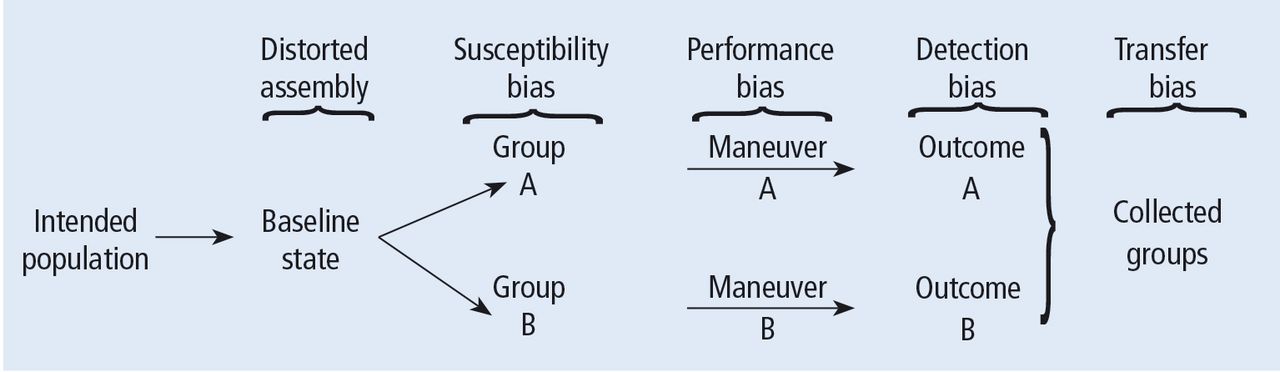
Potential sources of bias in a randomized, controlled trial according to Feinstein.1
Hopefully, you are already beginning to get a sense that one has to be extremely disciplined in thinking about each of these sources of bias because they can have some very subtle nuances in randomized trials that can easily escape attention.
Returning to sources of bias, let’s consider the second type of bias, “performance bias.” Performance bias relates to the administration of the compared maneuvers—the primary or principal maneuver, compared with the comparative maneuver. Performance bias can occur when the main maneuver is not administered adequately or when the co-maneuvers are administered in an imbalanced way between the compared groups. Consider the example of the Long-Term Oxygen Treatment Trial (LOTT) trial, which compared use of supplemental oxygen with no supplemental oxygen in patients with stable COPD and resting or exercise-induced moderate desaturation.9 The principal outcome measure of LOTT was all-cause hospitalization or death. In such a study, many potential sources of performance bias exist. For example, performance bias might exist if none of the patients allocated to oxygen actually used supplemental oxygen. Alternately, to the extent that use of inhaled corticosteroids or antimuscarinic agents lessens the risk of COPD exacerbation, performance bias could occur if use of these co-maneuvers was imbalanced between the compared groups. As a specific extreme circumstance, if all patients in the nonoxygen group used these inhalers but none of the patients in the oxygen group did, then a lack of difference between exacerbation frequency could be related to this imbalance in co-maneuvers (a form of performance bias) rather than to the lack of efficacy of supplemental oxygen.
“Compliance bias” is a subset of performance bias which occurs when 2 conditions are satisfied: (1) the main maneuver is not administered adequately, and (2) the investigator is unaware of that nonreceipt so that this cannot be accounted for in interpreting the study results. For example, if a drug has efficacy but if no one in the treatment arm of the trial takes the drug, the absence of a difference in outcomes between the compared groups will be ascribed to nonefficacy, whereas “compliance bias” (ie, no one actually took the drug) could actually be the cause. Ideally, randomized studies should be evaluated on an “intention to treat” basis irrespective of compliance, but there is an analytic approach called “per protocol” analysis in which you can analyze the results according to whether the patient actually used the intervention in an effective way. “Per protocol” analysis is a secondary analysis of the primary results but it can nonetheless help determine whether the negative result is likely related to noncompliance or not.
A third type of internal bias, “detection bias,” is fairly straightforward. Detection bias is related to how avidly and how comparably the outcomes are measured between the 2 compared groups. Let’s say that you are conducting a trial of a new antibiotic and the primary outcome is colony counts on petri dishes of plated collected specimens. If the technicians who read the petri dish counts are unblinded, they may look at the colony counts with a biased eye, seeing fewer colonies on plates collected from patients receiving the antibiotic.
Overall, detection bias occurs when outcomes are ascertained or detected unequally between the compared groups, and detection bias can involve any of the following: is there comparable surveillance of the 2 groups for analysis of the outcome measure? Are the diagnostic tests comparably performed in both groups and is the interpretation comparably unbiased with equipoise? Investigators who know which patients are receiving an active drug and those who are not could experience subliminal bias that renders them more likely to find that the drug under study is efficacious.
Depending on the principal study maneuver, ensuring blinding can be challenging. To demonstrate this point, let’s consider the example of conducting a randomized control trial of Vicks VapoRub. Vicks VapoRub is an old product that smells like wintergreen and that mothers used to rub on the chests of their infants in the hope of speeding recovery from colds and bronchitis episodes. It was felt that the distinctive smell of the product was materially related to wintergreen, which gives rise to the odor. So, imagine a randomized trial of Vicks VaporRub. A trial is designed in which sick children receive Vicks VapoRub on their chest and others receive a placebo rub that lacks the distinctive wintergreen odor. But, the odor itself is felt to be related to how Vicks VapoRub actually works. Thus, it is the odor itself that creates the blinding challenge here.
The primary outcomes in this study are the duration of the child’s cold symptoms, as ascertained by pediatricians actually examining the children. So, pediatricians would come and listen to the infants’ chests: “Yeah, this chest is clear, but this other infant is still full of rhonchi,” and they would ascertain the outcome measure in this way. So, my blinding question to you is: how do you blind a trial of Vicks VapoRub given the conditions described? Namely, you put the VapoRub on the chest, it smells and the smell is the intervention—how do you blind such a trial?
The clever answer is that you should put Vicks VapoRub on the upper lips of all the examiners, so what they smell is Vicks VapoRub independent of whether the child they are examining also has the Vicks VapoRub or placebo on their chest. In this way, single blinding of the examiners is preserved and detection bias is averted. It is important to point out that double blinding could also be achieved by placing Vicks VapoRub on the child’s upper lip, but there is little reason to suspect that the infants being studied have a bias related to whether they smell the Vicks VapoRub.
The fourth potential source of internal bias is called “transfer bias.” Transfer bias is the selective loss to follow-up of patients from 1 of the 2 compared groups in the trial for a systematic reason. By systematic, I mean that that the drop-out is associated with the development of the outcome event or some impact of the intervention regarding the likelihood to develop the outcome event. As an example, if all patients respond favorably to a drug and everybody fails to follow up because they feel too good to come back, then that would bias the study towards nonefficacy even in the face of an efficacious intervention.
Finally, let’s consider a source of bias that can affect the “external validity,” or the generalizability of the study results to populations other than that included in the study itself. Dr. Feinstein calls this 5th type of bias “assembly bias” (Table 1).1 Assembly bias occurs when the results of the study cannot be reliably applied to populations outside the study itself.
For example, if I screen patients during a study of digoxin for heart rate control in atrial fibrillation, I could establish whether the subject was compliant or not by checking his/her serum digoxin levels. Serum levels of 0 indicate that the patient has not taken the digoxin. If I include a run-in period for the trial—an interval before the actual study when I am assessing potential subjects’ eligibility to participate—and check serum digoxin levels to include only patients who are shown to be taking the drug, then I am screening for study inclusion on compliance. In this way, I will have assembled a population that is highly compliant so that I can truly assess whether digoxin has efficacy in controlling the heart rate in patients with atrial fibrillation. At the same time, this study population is not highly representative of the population of patients with atrial fibrillation at large, because we know that rates of drug noncompliances may be as high as 30% to 40%. So, culling a population with run-in periods on demonstrated compliance criteria may be very important to assess efficacy (ie, whether the drug works), but this design will trade off on the effectiveness of the drug (ie, which asks the question “does the drug work in actual practice?”). This is because, in the yin-yang between assessing efficacy and assessing effectiveness, the focus on assessing efficacy naturally undermines the ability to assess whether the drug works in real-world conditions.
As another example of potential assembly bias, let’s say you are studying an antihypertensive drug at a Veterans Administration (VA) hospital, where most veterans are men. But you are treating women in your practice and wonder whether the drug, which works in a predominately male population, will work in your female patients. So, there could be assembly bias in applying the results of a VA study to a non-VA predominantly female population.
Having now described the design of clinical trials and the major sources of bias, let’s apply this thinking to the earliest clinical trial. James Lind, a British Naval officer, was credited with conducting the first clinical trial of citrus fruits for scurvy while sailing on the ship Salisbury in 1747.2 The question that Lind addressed was “does citrus fruit treat and prevent scurvy?” In describing this trial, Lind stated “I took 12 patients with scurvy, these patients were as similar as I could have them, had one diet common to all.” As you read this through your new Feinsteinian bias lens, Lind is addressing 2 potential sources of bias, namely, susceptibility bias and performance bias. In trying to make the “cases as similar as I could have them,” he is trying to avoid susceptibility bias and in “providing one diet common to all,” he is trying to avoid performance bias.
In terms of the intervention in this trial, these 12 patients were allocated in pairs to several interventions: a quart of cider a day, 25 drops of elixir of vitriol 3 times a day on an empty stomach, 2 spoonsful of vinegar 3 times a day on an empty stomach, ½ pint a day of sea water, 2 oranges and 1 lemon given every day, and a “bigness of nutmeg” 3 times per day. In describing the outcome of the trial, Lind states “the consequence was that the most sudden and visible good effects were perceived from the use of oranges and lemons; one of those who had taken them, being at the end of 6 days fit for duty. The spots were not indeed at that time quite off his body, nor his gums sound, but without any other medicine then a gargarism of elixir vitriol, he became quite healthy before we came into Plymouth which was on the 16th of June. The other was the best recovered of any in his condition; and being now deemed pretty well, was appointed nurse to the rest of the sick.”
In analyzing this trial, we could characterize it as a parallel controlled trial. Whether the allocation was done by randomization is not clear, but it was certainly an observational cohort study in that there were concurrent controls who were treated as similarly as possible except for the principal maneuver, which was the administration of citrus fruit. Already mentioned was the attention to averting susceptibility and performance bias. There was no evidence of compliance bias as the interventions were enforced, nor was there evidence of transfer bias because all subjects who were enrolled in the study completed the study because they were a captive group on a sailing ship. Finally, the likelihood of assembly bias seems small, as these sailors seemed to be representative of victims of scurvy in general, namely in being otherwise deprived of access to citrus fruits.
In terms of the statistical results of this study, subsequent analysis of the research showed that the impact of lemons and oranges was dramatic and showed a trend (P = .09) towards statistical significance. Notwithstanding the lack of a P < .05, Dr. Feinstein would likely say that this study satisfied the “intra-ocular test” in that the efficacy of the citrus fruit was so dramatic that it “hit you between the eyes.” He often argued that the widespread practice of prescribing penicillin for pneumococcal pneumonia was not based on the results of a convincing randomized controlled trial because the efficacy of penicillin in that setting was so dramatic that a randomized trial was not necessary (and potentially even unethical if the condition of “intra-ocular” efficacy was satisfied).
The final question to address in this lecture is whether randomized controlled trials, for all their rigor, always produce more reliable results than observational studies. This issue has been addressed by several authors.10–12 Sacks et al10 contended in 1983 that observational studies systematically overestimate the magnitude of association between exposure and outcome and therefore argued that randomized trials were more reliable than observational studies. Subsequent analyses tended to challenge this view.11,12 Specifically, Benson and Hartz11 compared the results of 136 reports regarding 19 different therapies that were studied between 1985 and 1998. In only 2 of the 19 analyses did the treatment effects in the observational studies fall outside the 95% confidence interval for the randomized controlled trial results. In this way, these authors argued that observational studies generally are concordant with the results of randomized trials. They stated that “our finding that observational studies and randomized controlled trials usually produce similar results differs from the conclusions of previous authors. The fundamental criticism of observational studies is that unrecognized confounding factors may distort the results. According to the conventional wisdom, this distortion is sufficiently common and unpredictable that observational studies are not liable and should not be funded. Our results suggested observational studies usually do provide valid information.”11
An additional analysis of this issue was performed by Concato et al,12 who identified 99 articles regarding 5 clinical topics. Again, the results from randomized trials were compared with those of observational cohort or case-controlled studies regarding the same intervention. The authors reported that “contrary to prevailing belief, the average results from well-designed observational studies did not systematically overestimate the magnitude of the associations between exposure and outcome as compared with the results of randomized, controlled trials on the same topic. Rather, the summary results of randomized, controlled trials and observational studies were remarkably similar.”12
On the basis of these studies, it appears that randomized control trials continue to serve as the gold standard in clinical research, but we must also recognize that circumstances often preclude the conduct of a randomized trial. As an example, consider a randomized trial of whether cigarette smoking is harmful, which, given the strong suspicion of harm, would be unethical in that patients cannot be randomized to smoke. Similarly, from the example before, a randomized trial of penicillin for pneumococcal pneumonia would be unethical because denying patients in the placebo group access to penicillin would exclude them from access to a drug that has “intra-ocular” efficacy. In circumstances like these, well-performed observational studies that are attentive to sources of bias can likely produce comparably reliable results to randomized trials.
In the end, of course, the interpretation of the study results requires the reader’s careful attention to potential sources of bias that can compromise study validity. The hope is that with Dr. Feinstein’s framework, you can be better equipped to think critically about study results that you review and to keenly ascertain whether there is any threat to internal or to external validity. Similarly, as you go on to design clinical trials yourselves, you can pay attention to these potential sources of bias that, if present, can compromise the reliability of the study conclusions internally or their applicability to patients outside of the study.
Footnotes
This article is based on Dr. Stoller’s presentation at the “Biostatistics and Epidemiology” lecture series created by Aanchal Kapoor, MD, Critical Care Medicine, Cleveland Clinic. Dr. Stoller presented his lecture on August 2, 2016, at Cleveland Clinic.
Dr. Stoller reported research grant support from CSL Behring and consulting for Grifols, Shire, CSL Behring, and Arrowhead Pharmaceuticals.
- Copyright © 2017 The Cleveland Clinic Foundation. All Rights Reserved.
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.