


A 35-year-old woman with a medical history significant only for recently diagnosed essential hypertension presented to an urgent-care facility with easy bruising, petechiae, and gingival bleeding. She said that she had noticed large ecchymoses from minimal trauma on her upper and lower extremities for the past week, as well as a petechial rash. A complete blood cell count at that time revealed a platelet count of 3.0 × 109/L (reference range 150–400 × 109/L), and she was told to go to the nearest emergency department.
In the emergency department, repeat testing confirmed that her platelet count was indeed only 3.0 × 109/L, while the rest of her complete blood cell count values were unremarkable (Table 1). Her blood pressure was 167/104 mm Hg, heart rate 80 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 99% on room air.
The patient’s complete blood cell count
CAUSES OF THROMBOCYTOPENIA
1. All of the following are possible causes of thrombocytopenia except for which one?
Thrombotic thrombocytopenic purpura
Disseminated intravascular coagulation
Immune thrombocytopenic purpura
Glucose-6-phosphate dehydrogenase deficiency
Hemolytic uremic syndrome
The causes of thrombocytopenia can be divided into disorders of decreased platelet production and disorders of increased platelet consumption or destruction.
Disorders of decreased production include bone marrow diseases, such as myelodysplastic syndromes and aplastic anemia, and liver disease.
Disorders of platelet destruction or consumption include immune thrombocytopenic purpura and hemolytic processes. Thrombocytopenia can also occur during massive fluid resuscitation or blood transfusion without the transfusion of platelets, known as posttransfusion purpura, or during hypersplenism.1
Thrombotic thrombocytopenic purpura
Thrombotic thrombocytopenic purpura is caused by a genetic or acquired deficiency of ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13).
ADAMTS13 is a protease responsible for cleaving von Willebrand factor during times of high shear stress and resultant platelet adhesion, preventing large von Willebrand factor multimers from accumulating. A deficiency of ADAMTS13 leads to accumulation of large von Willebrand factor multimers that platelets then attach to, leading to widespread microvascular thrombosis.1,2
The clinical features seen with thrombotic thrombocytopenic purpura are widespread and may include symptoms of thrombocytopenia and hemolytic anemia; renal dysfunction; neurologic impairment, including headaches, confusion, or even stroke and seizures; gastrointestinal symptoms such as nausea, vomiting, diarrhea, and abdominal pain; and fever. Laboratory findings include those typically seen in hemolytic anemia, including low haptoglobin, increased lactate dehydrogenase, and an increased reticulocyte count, as well as thrombocytopenia, schistocytes on peripheral blood smear, and possibly increased creatinine with proteinuria, hematuria, or both.2
The PLASMIC score3 can be used to predict the likelihood of ADAMTS13 deficiency in patients with thrombocytopenia and schistocytes. It is calculated by awarding 1 point for each of the following features, if present:
Platelet count < 30 × 109/L
Hemolysis (reticulocyte count > 2.5%, undetectable haptoglobin, or indirect bilirubin > 2 mg/dL)
No active cancer
No solid organ or stem cell transplant
Mean corpuscular volume < 90 fL
International normalized ratio < 1.5
Creatinine < 2.0 mg/dL.
Patients with scores of 5 or higher should be empirically treated for thrombotic thrombocytopenic purpura with plasma exchange, as this condition can be life-threatening if untreated.4
Disseminated intravascular coagulation
Disseminated intravascular coagulation is a clinical syndrome in which the processes of both coagulation and fibrinolysis are inappropriately activated, leading to bleeding, clotting, or both. The coagulation and fibrinolytic processes may be inappropriately activated during sepsis or in other conditions such as pregnancy or malignancy. Typical laboratory findings include prolonged prothrombin time and partial thromboplastin time, low levels of fibrinogen, increased levels of D-dimer, and findings consistent with microangiopathic hemolytic anemias.5,6
Immune thrombocytopenic purpura
Immune thrombocytopenic purpura is an acquired, isolated thrombocytopenia modulated by platelet autoantibodies, with a platelet count less than 100 × 109/L.7 It is a diagnosis of exclusion, with clinical features of thrombocytopenia ranging from petechiae, mucosal bleeding, and easy bruising to internal bleeding and hemorrhagic stroke. Other than a low platelet count, laboratory data are typically normal.
Primary immune thrombocytopenic purpura is due to production of autoantibodies against platelets, whereas the secondary form can be triggered by many different conditions, including viral illnesses and autoimmune diseases.7,8
Glucose-6-phosphate dehydrogenase deficiency
Glucose-6 phosphate dehydrogenase deficiency is an X-linked genetic disorder that can lead to hemolytic anemia after ingestion of certain foods or drugs. Glucose-6 phosphate dehydrogenase is an enzyme that protects red blood cells from oxidative injury, and lack of it renders red blood cells susceptible to oxidative damage. As a result, red blood cells are lysed during times of oxidative stress.9
Laboratory data reveal a hemolytic process, with schistocytes and bite cells on peripheral blood smear, decreased levels of haptoglobin, and increased lactate dehydrogenase. However, platelet counts and coagulation studies are typically unaffected.10 Therefore, this is the correct answer to the question above.
Hemolytic uremic syndrome
Hemolytic uremic syndrome is a thrombotic microangiopathy that is caused by infection with Shiga toxin-producing Escherichia coli and leads to acute kidney injury, hemolytic anemia, and thrombocytopenia. Clinical features typically include abdominal pain and a diarrheal illness, and laboratory data reveals a hemolytic process with increased lactate dehydrogenase and decreased levels of haptoglobin, schistocytes on peripheral blood smear, thrombocytopenia, normal ADAMTS13 levels, normal prothrombin time and partial thromboplastin time, and possibly rising creatinine, hematuria, proteinuria, and hypertension.11
The laboratory findings commonly seen in thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, disseminated intravascular coagulation, and immune thrombocytopenic purpura are summarized in Table 2.
Differential diagnosis for thrombocytopenia
BACK TO THE PATIENT
The patient’s complete blood cell count (Table 1), chemistry panel (Table 3), and other tests (Table 4) excluded other differential diagnostic considerations, including thrombotic thrombocytopenic purpura, disseminated intravascular coagulation, and other hemolytic processes. She had not received any transfusions (which would have suggested posttransfusion purpura), and she had not received any medications commonly associated with drug-induced thrombocytopenic purpura, such as beta-lactam antibiotics. A peripheral blood smear revealed thrombocytopenia and normal-appearing white and red blood cells, indicative of immune thrombocytopenic purpura (Figure 1).
The patient’s chemistry panel
The patient’s other blood tests
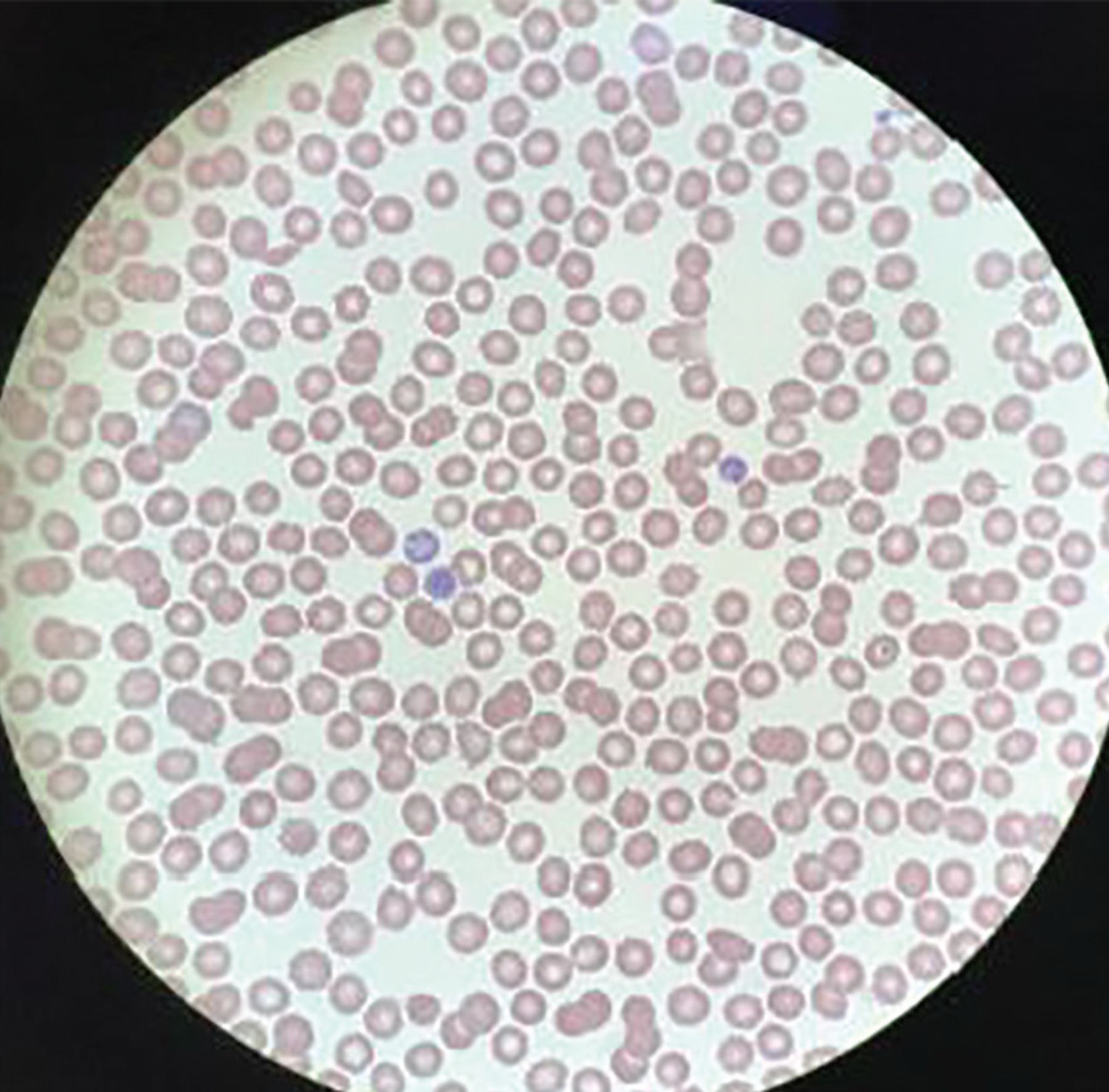
On the patient’s peripheral blood smear, no platelets were visible.
CAUSES OF SECONDARY IMMUNE THROMBOCYTOPENIC PURPURA
2. All of the following are causes of secondary immune thrombocytopenic purpura except which one?
Human immunodeficiency virus
Chronic obstructive pulmonary disease
Systemic lupus erythematosus
Hepatitis
Chronic lymphocytic leukemia
Viral infections are often implicated in secondary immune thrombocytopenia, including hepatitis C, human immunodeficiency virus, many herpesviridae such as Epstein-Barr virus and cytomegalovirus, and others.12,13 Autoimmune disorders such as systemic lupus erythematosus, rheumatoid arthritis, and hematologic malignancies can be implicated in immune thrombocytopenic purpura as well.
Chronic obstructive pulmonary disease, on the other hand, has not been documented as a cause of secondary immune thrombocytopenic purpura, although it can cause a reactive polycythemia as a result of chronic hypoxia.14 Therefore chronic obstructive pulmonary disease is the correct answer.
AN INTERESTING AND TIMELY PLOT TWIST
A nasal swab for SARS-CoV-2—the cause of coronavirus disease 2019 (COVID-19)—was ordered, as is currently standard practice for all of our admitted patients, and her test was positive by polymerase chain reaction assay. The patient said she had no shortness of breath, cough, fevers or chills, diarrhea, anosmia, or dysgeusia on admission or in the past weeks or months. She did not need supplemental oxygen or intensive care. She said she had been compliant with mask-wearing and social distancing, but she worked for a cleaning company and had been in contact with many people and potentially contaminated surfaces.
In view of the pertinent negative findings described above and the temporal relationship between her symptoms and her positive SARS-CoV-2 test, we concluded that her otherwise-asymptomatic COVID-19 was the trigger for her severe thrombocytopenia.
TREATMENT OF SEVERE SECONDARY IMMUNE THROMBOCYTOPENIA
3. All of the following are treatment options for severe secondary immune thrombocytopenic purpura except which one?
Glucocorticoids
Intravenous immunoglobulin
Rituximab
Splenectomy
Plasma exchange
First-line treatment for severe secondary immune thrombocytopenia includes glucocorticoids, intravenous immunoglobulin, or both. Both are thought to interfere with destruction of platelets.15 An additional first-line treatment is anti-D immunoglobulin. Second-line treatments include thrombopoietin receptor agonists, splenectomy, and rituximab.16
Plasma exchange is considered a first-line treatment for thrombotic thrombocytopenic purpura and serves to replace the deficient ADAMTS13 molecule. However, it is not typically used to treat immune thrombocytopenic purpura,16 and therefore this is the correct answer.
BACK TO THE PATIENT
In the emergency department, the patient received 1 dose of dexamethasone 40 mg and 2 units of platelets. She was admitted to the hospital for 2 days, during which she received 2 doses of oral prednisone 1 mg/kg/day. She was discharged home on the third day with instructions to take an additional 2 doses of prednisone 1 mg/kg/day. A repeat complete blood count after she finished her course of steroids, 5 days after her initial presentation, revealed a platelet count of 268 × 109/L.
HEMATOLOGIC COMPLICATIONS OF COVID-19
The thrombotic complications of COVID-19 have been well documented.17–20 Thrombocytopenia can occur with COVID-19 by various mechanisms, including disseminated intravascular coagulation and sepsis.21
In addition, there have been multiple case reports of COVID-19–induced severe secondary thrombocytopenia (Table 5).22–28 The patients all had typical symptoms of COVID-19 such as cough, fever, or shortness of breath. The timing of thrombocytopenia varied, with some patients developing it early in their hospital course and others developing it days after admission. All patients, excluding 1 who died shortly after developing thrombocytopenia, were treated with intravenous immunoglobulin, corticosteroids, or both, with hematologic recovery in all reported cases. To our knowledge, however, ours is the first documented case of a SARS-CoV-2–positive patient presenting with symptomatic severe thrombocytopenia but no COVID-19 symptoms.
Reported cases of COVID-19-associated immune thrombocytopenic purpura
This patient’s experience further reveals that SARS-CoV-2 can cause severe secondary immune thrombocytopenia, and is unique in showing that thrombocytopenia can be the sole presenting disorder in COVID-19. Hematologic monitoring of COVID-19 patients is becoming increasingly important, especially with respect to hypercoagulable complications,29 but attention must also be directed to platelet counts. Conversely, patients presenting with isolated thrombocytopenia should be screened for SARS-CoV-2 infection.
DISCLOSURES
The authors report no relevant financial relationships which, in the context of their contributions, could be perceived as a potential conflict of interest.
- Copyright © 2021 The Cleveland Clinic Foundation. All Rights Reserved.
REFERENCES
In this issue




{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.