


ABSTRACT
Antibody-mediated autoimmune encephalitis (AE) is a heterogeneous group of inflammatory central nervous system disorders. Symptoms typically include subacute, progressive neuropsychiatric symptoms with associated cognitive dysfunction, movement disorders, and autoimmune seizures. The diagnosis should be based on objective neurologic dysfunction in combination with auto antibody testing. Treatment with immunotherapies requires both short-term and long-term strategies depending on the specific syndrome and potential for relapse. In this paper, we review key features of AE, focusing on syndromes involving cell surface and synaptic proteins, and share a practical approach to the diagnosis and management, including common pitfalls associated with nonspecific antibody findings.
AE is an umbrella term for a group of inflammatory central nervous system disorders associated with neuronal autoantibodies or other biomarkers of central nervous system autoimmunity.
Common clinical presentations include progressive neurocognitive symptoms with concomitant movement disorders, seizures, and autonomic dysfunction that worsens over weeks to months.
Objective clinical findings are needed to make the diagnosis of AE, including changes on magnetic resonance imaging, electroencephalography, and cerebrospinal fluid analysis.
The spectrum and understanding of antibody-mediated autoimmune encephalitis (AE)—an umbrella term for a group of noninfectious, inflammatory central nervous system diseases—have expanded dramatically over the past few years. Familiarity with AE syndromes ensures prompt diagnosis and treatment. Practitioners need to stay abreast of developments in this field as the breadth of immune-mediated disorders of the nervous system continues to evolve.
In this paper, we will focus on the clinical features of common central nervous system cell surface and synaptic antibody syndromes in adults and on the emerging evidence in this area that has led to rapid changes in management and treatment over the past decade. We will also briefly comment on antibody-negative AE.
Antibody-related syndromes such as intracellular neuronal antibody-associated encephalitis and encephalitis occurring with demyelinating disorders are less commonly encountered in clinical practice and are beyond the scope of this article.
GENERAL FEATURES OF AUTOIMMUNE ENCEPHALITIS
Potential associations of AE encompass paraneoplastic, parainfectious triggers along with adverse events related to various immunotherapies.1–3
Onset of AE is usually subacute over weeks to months, with progressive neurocognitive symptoms including encephalopathy, cognitive dysfunction, neuropsychiatric symptoms, and seizures. Other features may include brain-stem syndromes, dysautonomia, and movement disorders.
The prevalence and incidence are increasing as testing becomes more widely available. A recent study in Olmsted County, MN, showed a prevalence of AE of 13.7 per 100,000, similar to that of infectious encephalitis.4
HOW IS AUTOIMMUNE ENCEPHALITIS CLASSIFIED?
Over the past few decades, there has been rapid growth in the discovery of antibody-associated neurologic diseases. Autoantibodies that target neuronal antigens can cause a diverse set of neurologic disorders. This has significantly raised awareness of the wide spectrum of disease presentations that may have an underlying autoimmune component.
Antibodies associated with AE are commonly divided into 2 groups depending on the location of the antigen. The traditional “well-defined” syndromes (eg, anti-Hu or ANNA-1, anti-Ri or ANNA-2) target intracellular neuronal antigens.5 And more recently, a new group of neuronal cell surface/synaptic proteins has been described in association with AE. This distinction is important for diagnosis and prognosis. Intracellular antibody-mediated syndromes appear to be driven primarily by a CD8+ T-cell cytotoxic response and usually have a poorer prognosis, with a limited response to immunotherapy (Table 1). In contrast, cell surface/synaptic antibodies appear to be directly pathogenic and are more responsive to multimodal immunotherapies (Table 2).1,5 Detailed discussions of the pathophysiology of AE have been published by Bradshaw and Linnoila6 and by McKeon.7
Autoantibody biomarkers of autoimmune encephalitis: Intracellular autoantibodies
Autoantibody biomarkers of autoimmune encephalitis: Cell-surface and synaptic antibodies
Though the terms paraneoplastic syndrome and AE are sometimes used interchangeably, not all AE syndromes are paraneoplastic. Paraneoplastic syndromes are defined as neurologic syndromes occurring in the setting of cancer, sometimes preceding the diagnosis of neoplasm by months or years.1 Paraneoplastic disorders are usually related to intracellular neuronal antibodies. Cell surface/synaptic antibody-mediated syndromes, however, may also be associated with cancer, though they are usually classified as phenotypes of low to moderate risk, as these disorders can occur with or without cancer. The strength of association with an underlying neoplasm varies depending on the specific antibody or antibodies.5
WHEN SHOULD I CONSIDER THE DIAGNOSIS?
AE may be considered in patients presenting with subacute onset (over 1 to 3 months) of cognitive or memory deficits, alterations in consciousness, seizures, movement disorders, or other neuropsychiatric symptoms.8 Accompanying neurologic or systemic symptoms suggestive of a specific antibody-mediated syndrome can increase clinical suspicion for AE. Examples include the following:
Dystonia-chorea for anti-N-methyl-D-aspartate (anti-NMDA) receptor encephalitis
Hyperekplexia (exaggerated startle reflex) for anti-glycine receptor (Gly-R) antibody syndrome
Faciobrachial dystonic seizure (focal or lateralized coordinated contractions of an arm and the face) for antileucine-rich glioma-inactivated protein 1 (LGI1) encephalitis
Peripheral nerve hyper excitability (diffuse involuntary motor-unit activity due to hyperexcitability of the motor nerve or its terminal)9 for anti-contactin-associated protein-like 2 (Caspr2) syndrome
Weight loss accompanying gastrointestinal symptoms for anti-dipeptidyl-peptidase-like protein 6 (DPPX) encephalitis.5,10
While certain autoantibody disorders have a specific phenotype, a number of patients with AE do not present with classic “limbic encephalitis” and can present with a range of central nervous system and peripheral nervous system involvement.11 Table 3 provides a list of potential clinical and radiographic findings suggestive of AE.
Clinical, diagnostic, and radiographic clues to autoimmune encephalitis
It is also important to recognize that AE can be antibody-negative. Antibody-negative AE may occur due to limitations of currently available testing, especially as novel autoantibodies are being discovered. However, objective clinical and radiologic criteria exist to aid diagnosis.8
Despite the challenges, the diagnosis of AE should be driven by the patient’s clinical presentation and diagnostic evaluation. This approach includes a detailed clinical history including a personal or family history of autoimmunity, identifying infectious risk factors (ie, exposure and travel history) while excluding other conditions in the differential diagnosis.8 It should also be noted that a patient with AE may not exhibit all the disease characteristics discussed below. For example, normal findings on magnetic resonance imaging (MRI) and electroencephalography (EEG) are not uncommon in anti-LGI1 encephalitis.12
WHAT ARE THE COMMON CELL-SURFACE/SYNAPTIC ANTIBODY SYNDROMES IN AUTOIMMUNE ENCEPHALITIS?
Anti-NMDA receptor encephalitis
Anti-NMDA receptor encephalitis was initially characterized in 12 women showing a characteristic progression of psychiatric symptoms ranging from subtle behavioral changes, such as irritability, to frank psychosis. These symptoms were followed by movement disorders, autonomic dysfunction, hypoventilation, seizures, and coma.13 Anti-NMDA is one of the most frequently identified neuronal autoantibodies in AE.1 Anti-NMDA receptor encephalitis frequently affects young adults, with a strong female predominance (4:1), though it has been described in all ages including children and the elderly, with variable phenotypes.14 The median age is 20.
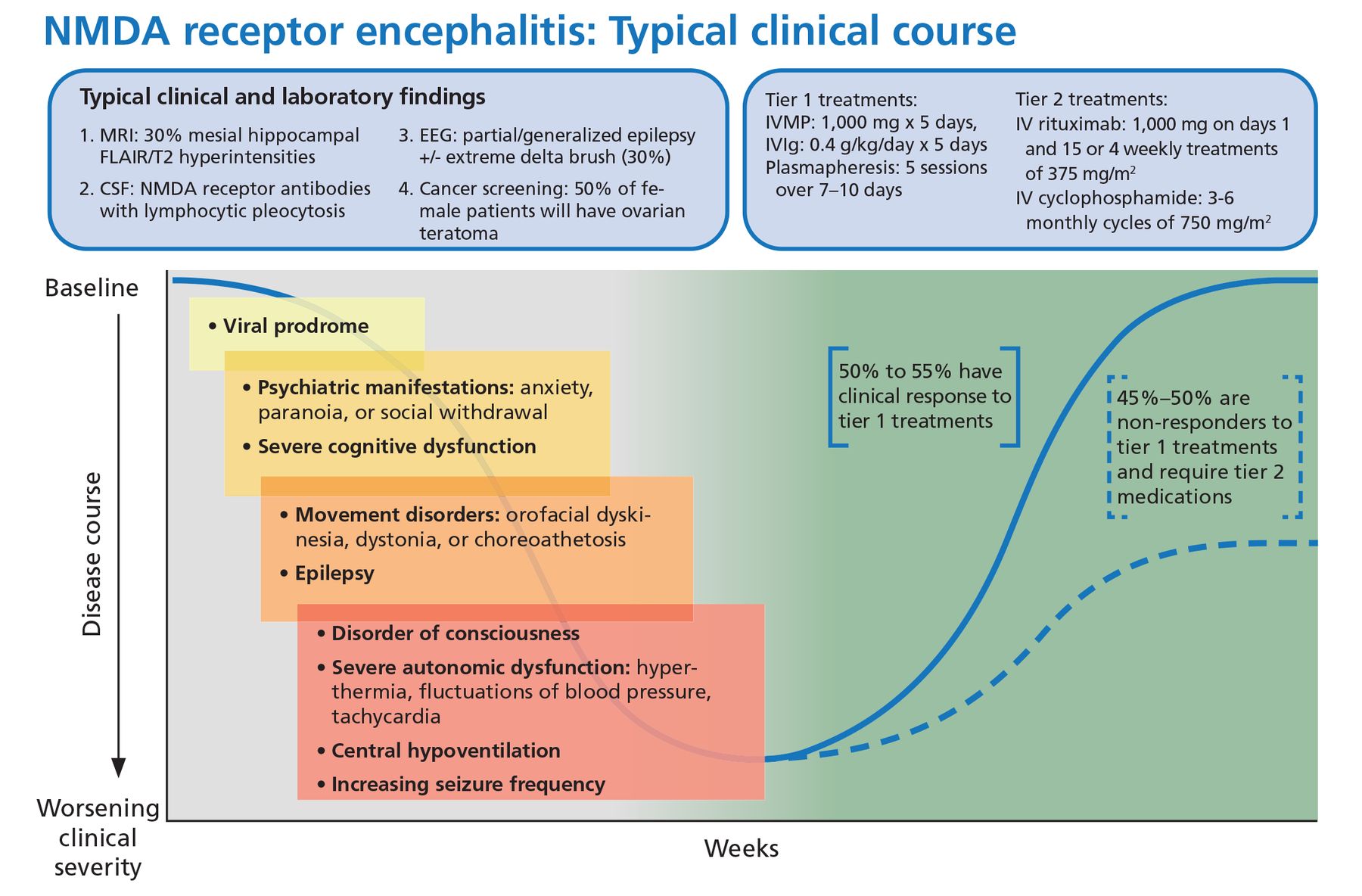
Viral-like prodrome. Some patients experience a viral-like prodrome that includes headache or fever during the initial 1 to 2 weeks of the illness (Figure 1). Soon after, subacute psychiatric symptoms develop including anxiety, personality changes, hallucinations, paranoid ideas, and frank psychosis. It is not unusual for patients to alternate between hyperactive and catatonic states. Concomitant movement disorders such as dyskinesia (typically orofacial or limb), dystonia, and choreoathetosis are common. From 60% to 75% of adult patients have been reported to experience behavioral problems or movement disorders during the first month of the disorder.15
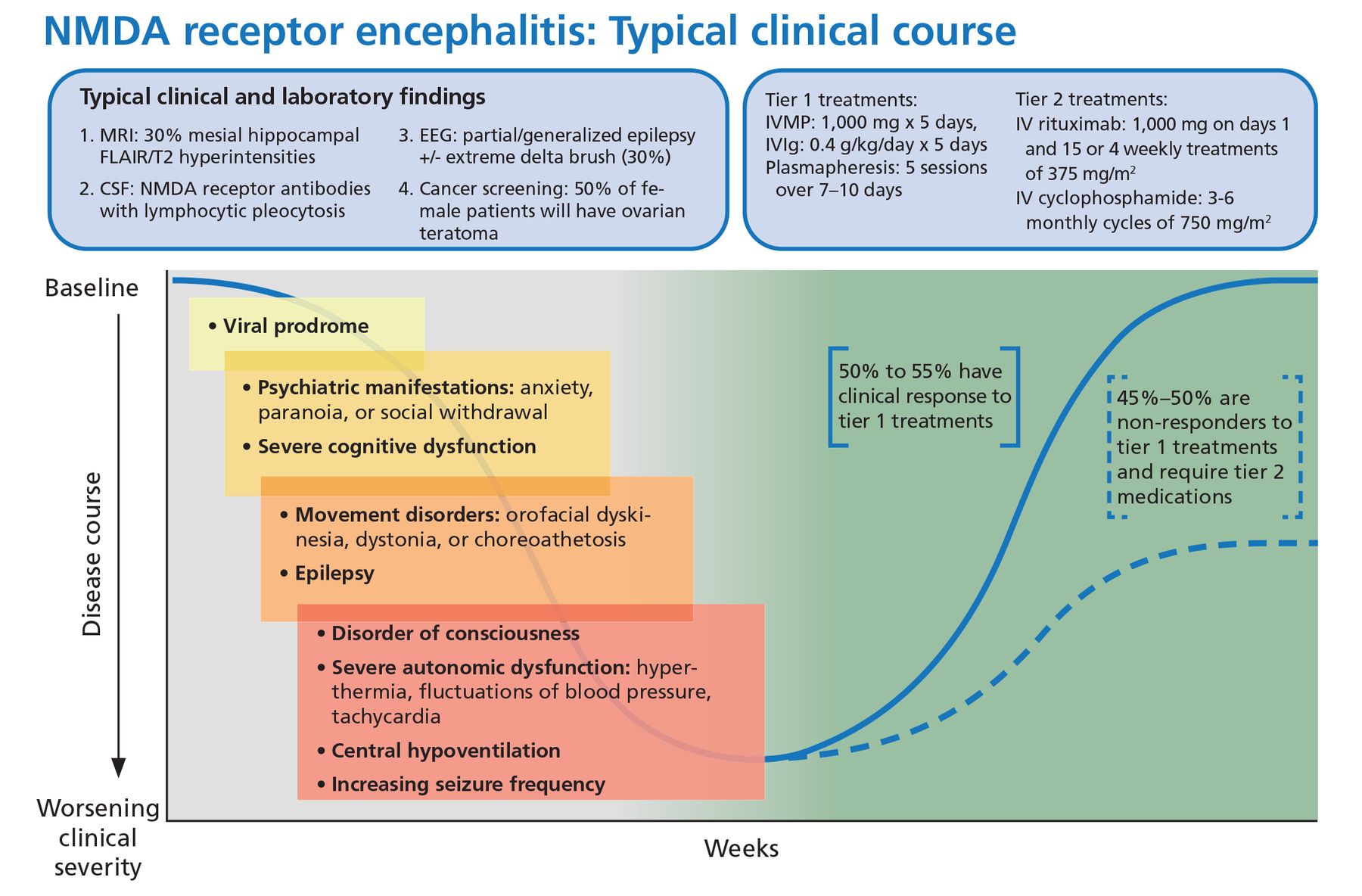
Typical clinical course associated with anti-NMDA receptor encephalitis.
CSF = cerebrospinal fluid; EEG = electroencephalography; FLAIR = fluid-attenuated inversion recovery; IV = intravenous; IVIg = intravenous immunoglobulin; IVMP = intravenous methylprednisolone; MRI = magnetic resonance imaging; NMDA = anti-N-methyl-D-aspartate receptor
Based on information in reference 15.
Autonomic dysfunction. As the disease progresses, autonomic dysfunction becomes more prominent and commonly necessitates monitoring in an intensive care unit.
Potential complications include tachyarrhythmias, hypotension, and central hypoventilation requiring mechanical ventilation. Approximately 80% of patients require ICU admission.15
Seizures can occur at any time and are often not responsive to antiepileptic medications alone. In a case series of 75 patients with anti-NMDA receptor encephalitis, almost 80% suffered from generalized tonicclonic seizures, and 74% had focal seizures without impaired awareness.16 There has been some evidence to suggest antiepileptic drugs with sodium channel-blocking properties (eg, carbamazepine, oxcarbazepine, lacosamide, phenytoin) may be more effective, though seizure freedom is usually achieved only when paired with immunotherapies.16,17
NMDA receptor immunoglobulin G
(IgG) testing should always be done with cerebrospinal fluid (CSF), as serum testing is less reliable (100% sensitivity for CSF vs 85% for serum).15 CSF analysis usually reveals a moderate lymphocytic pleocytosis, elevated protein, and intrathecal antibody production.
Patterns on EEG can vary and often simply demonstrate slowing.11 Approximately one-third of patients develop extreme delta brush; this is described as slowing with delta activity 1–3 Hz and superimposed bursts of rhythmic activity (beta activity 20–30 Hz) on these slow waves and may be a poor prognostic sign.18
MRI findings. MRI is usually normal but may show subtle mesial hippocampal fluid-attenuated inversion recovery (FLAIR) hyperintensities. Titulaer et al reported that 76% of patients had a CSF pleocytosis, one-third of the cohort had an abnormal brain MRI, and 90% had changes on EEG including slowing.15
Teratomas. Anti-NMDA receptor encephalitis may be associated with ovarian or extraovarian teratomas in 30% to 40% of patients, particularly in young women. It requires prompt surgical treatment.15
Infection. Herpes simplex virus infection of the central nervous system can also trigger the production of NMDA receptor IgG.19 In a large prospective study, 27% of patients with herpes simplex encephalitis developed anti-NMDA receptor antibodies within 16 weeks of completing their acyclovir treatment.19 None of the patients had NMDA receptor IgG at the index admission. Interestingly, 3 patients (6%) developed CSF NMDA receptor IgG without clinical correlation, but the antibody production was not detectable at 1-year follow-up.19 There have been reports of pediatric anti-NMDA receptor encephalitis developing after Japanese encephalitis infection, but no other clear postinfectious or vaccination pattern has emerged in the literature.20,21
Immunotherapy. Anti-NMDA receptor encephalitis responds to immunotherapy, but the response can be slow. In the largest study to date of patients with anti-NMDA receptor encephalitis, 81% had significant recovery at 24 months, but only 53% had clinical improvement within 4 weeks of diagnosis.15
From 10% to 25% of patients have a clinical relapse, though symptoms are less severe than in the initial presentation.1,22
Maintenance immunotherapy can be used to optimize acute treatment response while preventing relapses.15 Commonly used agents include oral corticosteroids, intravenous immunoglobulin (IVIg), and steroid-sparing agents such as mycophenolate mofetil, azathioprine, rituximab, and cyclophosphamide.23
Anti-LGI1 encephalitis
Anti-LGI1 encephalitis was first characterized in 2010.24 In contrast to anti-NMDA receptor encephalitis, it typically occurs in men over age 60, presenting with subacute cognitive dysfunction and behavioral changes.
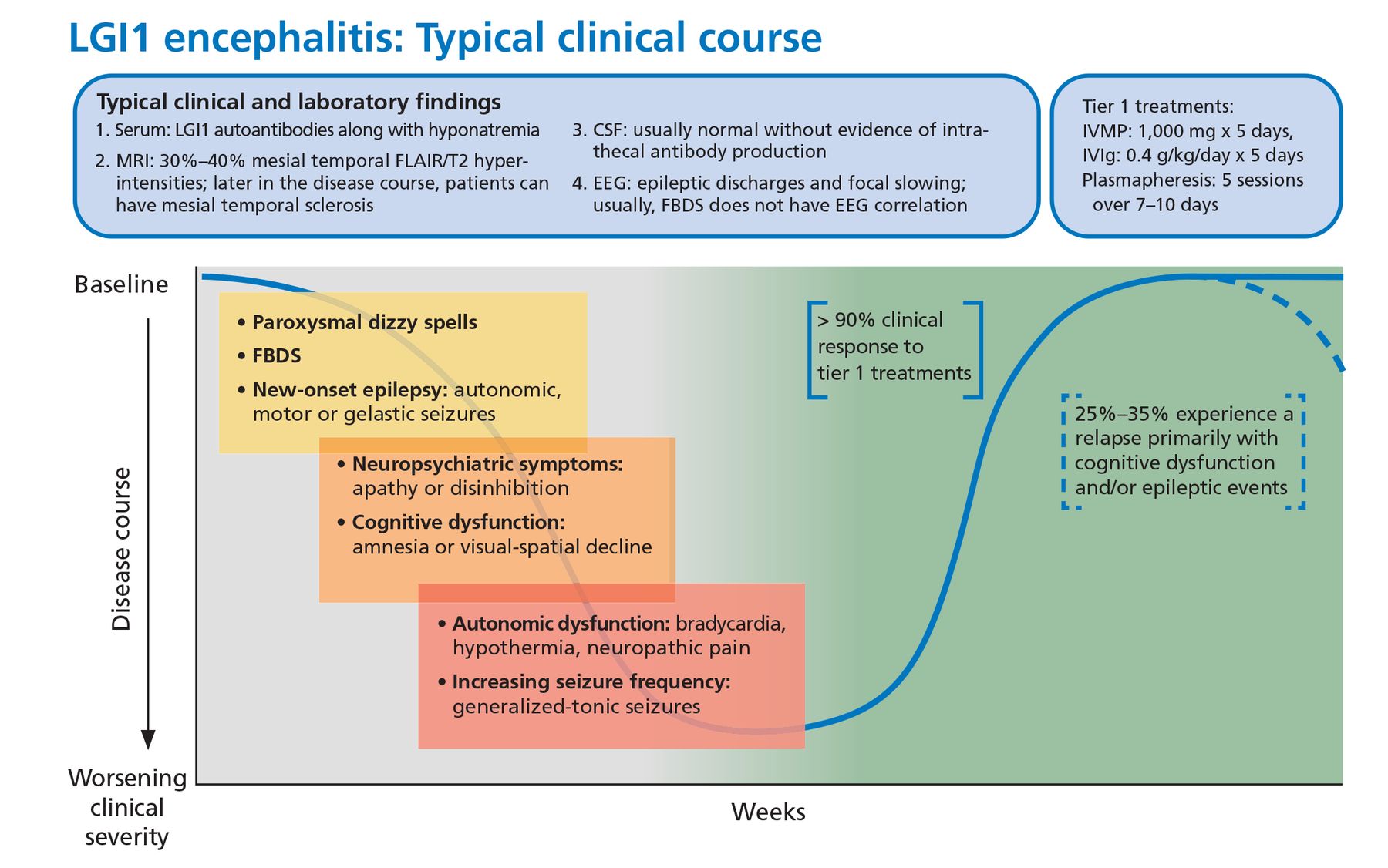
About 50% of patients also develop faciobrachial dystonic seizures characterized by focal or lateralized rapid coordinated movements of an arm or the face that may occur hundreds of times a day. These dystonic seizures are very specific for anti-LGI1 encephalitis but are not universally present. Most patients will have normal surface activity on EEG even if the faciobrachial seizure events are captured during the recording. The exact reason for this is unclear, but it may reflect subcortical seizure origin, as some of these patients have contralateral basal ganglia lesions on MRI.25
Other associated seizure subtypes include subtle autonomic focal seizures and generalized tonic-clonic seizures (Figure 2).12 Autonomic hyperactivity has also been reported. This includes hyperhidrosis, tachycardia, blood pressure lability, and urinary dysfunction.26
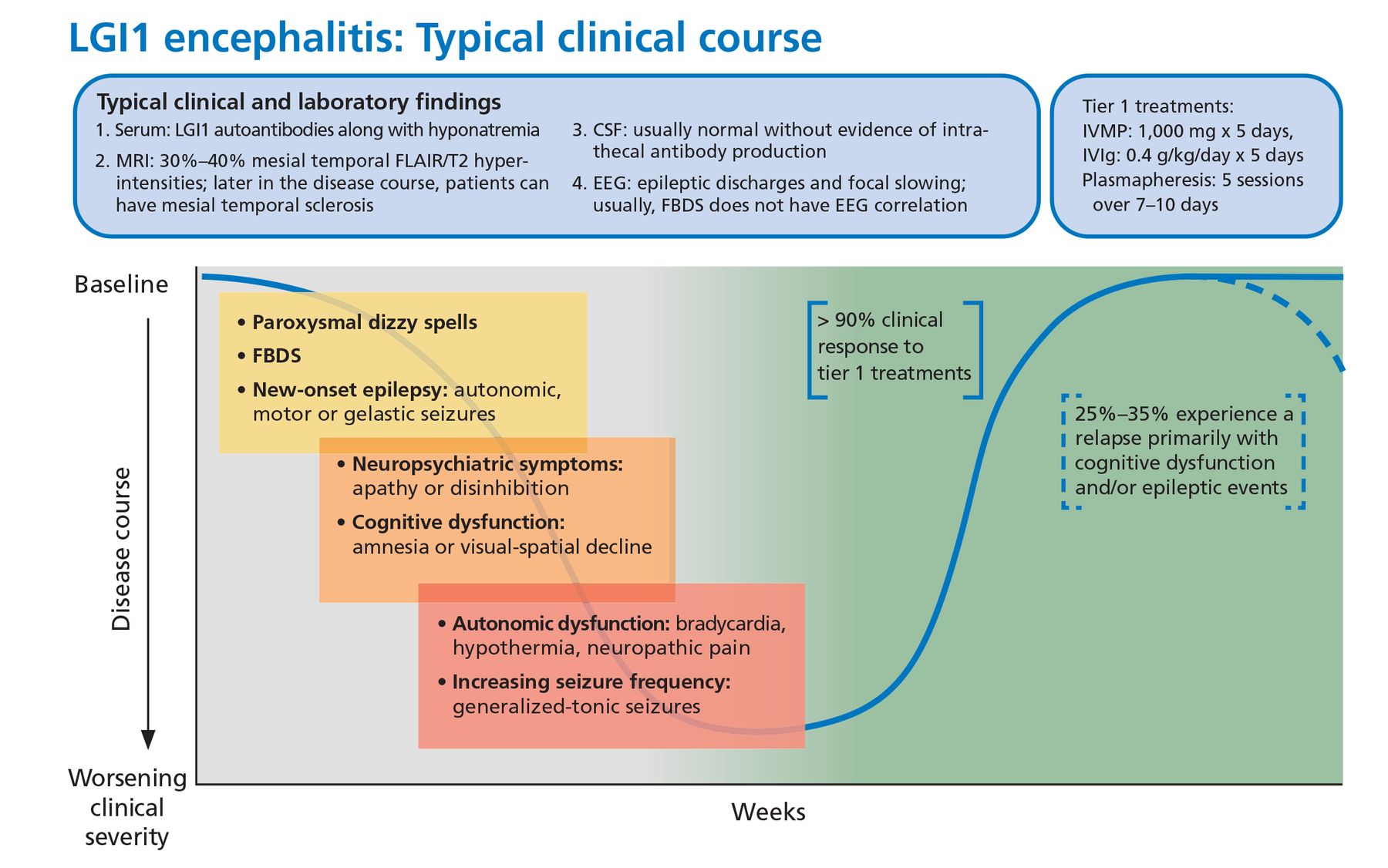
Typical clinical course associated with anti-LGI1 encephalitis.
CSF = cerebrospinal fluid; EEG = electroencephalography; FBDS = faciobrachial dystonic seizures; FLAIR = fluid-attenuated inversion recovery; IVIg = intravenous immunoglobulin; IVMP = intravenous methylprednisolone; LGI1 = leucine-rich glioma-inactivated 1; MRI = magnetic resonance imaging
The initial workup for anti-LGI1 encephalitis includes MRI, EEG, and CSF analysis but is usually unrevealing aside from a mild hyponatremia on basic metabolic testing. CSF-specific oligoclonal bands may be seen, but CSF can also be non-inflammatory. Initial brain MRI can occasionally demonstrate T2-FLAIR hyperintensity of the hippocampus.27 Unlike in anti-NMDA receptor encephalitis, CSF is less sensitive than serum for the detection of LGI1 antibodies (serum 100% vs CSF about 88%).27,28
Treatment response. This syndrome characteristically responds briskly to tier 1 treatments (corticosteroids, intravenous immunoglobulin, plasmapheresis), but 20% to 30% of patients may experience a relapse necessitating long-term immunosuppression.22 In one small randomized control trial, IVIg treatment was associated with a significant reduction in seizure burden and can be considered a steroid-sparing agent.29
Malignancy. From 5% to 15% of patients have an underlying malignancy, most commonly thymoma.30 Patients may have residual cognitive deficits despite initial recovery.
WHAT TESTING SHOULD I CONSIDER FOR AE DIAGNOSIS?
When AE is highly suspected (Table 3), initial testing should include an antibody panel. Both serum and CSF should be tested for antibodies since CSF is more sensitive and specific for certain antibodies such as NMDA receptor IgG, GAD65 IgG, and GFAP IgG, whereas serum is more sensitive for other antibodies such as LGI1 IgG and Caspr2 IgG.
Antibody panels are preferred over specific antibody tests, given the often overlapping clinical syndromes and the possibility of multiple positive antibodies.
Common testing sites include Mayo Clinic or Associated Regional and University Pathologists (ARUP) laboratory. Antibody panels include:
Mayo ENS-2 panel in serum (www.mayocliniclabs.com/test-catalog/Overview/92116)
Mayo ENC-2 panel in CSF (www.mayocliniclabs.com/test-catalog/Overview/92117)
ARUP Autoimmune Encephalitis Extended Panel in serum (https://ltd.aruplab.com/Tests/Pub/3001431).
Encephalitis has a broad differential diagnosis
The differential diagnosis for encephalitis is very broad and includes infections, toxic-metabolic encephalopathy, mitochondrial disorders, nutritional deficiencies, vascular disorders, malignancy, and demyelinating disorders. Clinicians should be particularly concerned about ruling out an infectious process given the immunotherapies utilized to treat AE. Infections to consider include herpes simplex virus encephalitis, human herpesvirus 6, human immunodeficiency virus, fungal infection (eg, cryptococcal), mycobacterial infection, Whipple disease, and neurosyphilis.
In general, viral infections usually cause a more profound CSF pleocytosis (a white blood cell count of 50–100/μL). Bacterial or mycobacterial infections can have a lower of CSF glucose concentration, whereas AE usually has normal glucose levels.31
Overall, careful examination usually reveals subtle neurologic deficits that should prompt further evaluation for AE. Diagnostic red flags include newly occurring epileptic seizures, movement disorders, and neurocognitive symptoms, especially in the setting of MRI or CSF abnormalities.
It should be mentioned that the prevalence of primary psychiatric disorders is much higher than the prevalence of AE. For example, the overall prevalence of schizophrenia, with incidence peaking in young adults, is estimated to be 2.7 to 8.3 per 1,000,32 whereas the overall prevalence of AE is 13.7 per 100,000.4 Thus, in a young patient with new mood disorder, a primary psychiatric diagnosis remains more likely than AE.
While patients with a preexisting psychiatric disorder can develop a concomitant autoimmune condition, it should also be mentioned that a specific isolated psychiatric AE phenotype has not emerged in the literature despite extensive investigations.21
A COMPREHENSIVE EVALUATION FOR AUTOIMMUNE ENCEPHALITIS
A comprehensive evaluation for suspected AE includes a range of laboratory studies and imaging.
Serum testing
Serum testing with an AE antibody panel to human immunodeficiency virus, thyroid-stimulating hormone, vitamin deficiency (B1, B12, E, folic acid).
Evaluation for other disease markers
Patients with autoimmune encephalitis are at increased risk of having a second autoimmune disorder. Choice of any specific testing should be based on clinical suspicion.7,33
Cerebrospinal fluid testing
CSF studies include an AE antibody panel, routine studies, CSF IgG index, CSF-specific oligoclonal bands, and comprehensive infectious evaluations with specific attention to viral agents (ie, herpes simplex virus type 1, varicella-zoster virus, West Nile virus, John Cunningham virus, and human herpesvirus 6).
Urine toxicology screen
A urine toxicology test should include screening for marijuana and cocaine.
Magnetic resonance imaging
MRI of the brain with and without gadolinium contrast is appropriate. Consider completing an epilepsy protocol, which may vary between institutions but usually consists of standard T1- and T2-weighted images, FLAIR, gradient-echo (T2) sequences, and diffusion-weighted sequences. Imaging sequences should also include contiguous, thin slices to ensure that hippocampal and temporal lobe lesions can be identified.34
Consider spinal cord MRI if neurologic abnormalities suggest concomitant myelitis. Symptoms could encompass motor, sensory, and autonomic (bladder, bowel, sexual) dysfunction that localizes to the spinal cord. An example could be a well-defined truncal sensory level below which the sensation is altered.35
Electroencephalographic monitoring
Continuous monitoring with EEG can clarify the cause of motor symptoms, identify suggestive patterns such as extreme delta brush, and characterize seizure burden.
Positron emission tomography
Brain fluorodeoxyglucose-positron emission tomography with computed tomography (FDG-PET/CT) may illustrate either hypo-metabolism that may correlate with impairment of neuronal activity even in the absence of structural disturbance36 or hypermetabolism that could correlate with increased glucose metabolism caused by synaptic dysfunction or ongoing seizure activity.37 Overall, PET may be a sensitive marker for AE but is limited by its higher cost, lack of diagnostic specificity and clinical availability.36
Body FDG-PET/CT can be done to screen for malignancy.38
WHICH AUTOANTIBODY FINDINGS SHOULD BE INTERPRETED CAUTIOUSLY?
The development of broad antibody panels has led to some unintended consequences, particularly as these panels may include disorders of the central nervous system and the peripheral nervous system.39 In addition, the specificity and sensitivity can vary for the different autoantibodies and the association with AE. Therefore, interpretation of autoantibody testing should be combined with a comprehensive clinical evaluation as outlined above in the section “A comprehensive evaluation for autoimmune encephalitis.”8
Caution is particularly needed when interpreting tests for autoantibodies that have low specificity for AE. For example, anti-voltage-gated potassium channel (VGKC) antibodies were initially detected in peripheral nerve hyperexcitability disorders such as neuro myotonia and Morvan syndrome. Further laboratory evaluation has elucidated that LGI1 and Caspr2 are the usual targets of these complex antibodies and not the VGKC itself. VGKC antibodies alone are not specific for AE and can be seen in 5% of healthy controls.40 In VGKC-positive patients without LGI1 and Caspr2 antibodies, additional testing is not usually warranted, as there is no clear evidence that VGKC titers are indicative of an autoimmune disorder.30
Hashimoto encephalopathy, also known as steroid-responsive encephalopathy associated with autoimmune thyroiditis, has been historically linked to elevated serum levels of thyroid peroxidase antibodies.41 In the era of more neuronal-specific antibodies, thyroid peroxidase antibodies have been found to have limited diagnostic value. An extensive evaluation to exclude other causes of AE should be pursued in cases with elevated thyroid peroxidase antibody titers, given the unclear clinical significance of these antibodies in neurologic disorders.42,43
WHAT IS THE INITIAL TREATMENT FOR AUTOIMMUNE ENCEPHALITIS?
Current treatment guidelines for AE are based on a combination of expert opinion, case series, and case reports, and evidence from high-quality multicenter randomized trials is lacking.29 But existing evidence suggests that early initiation of therapy and prompt escalation to second-line immunotherapy may lead to improved clinical outcomes.15,22
Still, unanswered questions include the time frame associated with a response to the first-line immunotherapy and the optimal duration of sustained immunotherapy. A comprehensive evaluation for malignancy is also vital as early detection and treatment are important for improved patient outcomes.
An important caveat for all clinicians is that although response to corticosteroid therapy is a typical feature of AE, it does not confirm the diagnosis of AE. Disorders such as lymphoma and vasogenic edema associated with a brain tumor can also respond dramatically to steroids. Additionally, some AE patients do not respond to corticosteroids but require prolonged treatment with other immunotherapies.8
Initiation and escalation of treatment depend on the pretest probability of AE along with the clinical severity. If there is a high pretest probability of a known syndrome, waiting for the results of antibody testing should not delay treatment. For example, a case of refractory autoimmune-mediated status epilepticus in the intensive care unit requires prompt and aggressive treatment. Caution should be used when the diagnosis is less certain.8
First-line treatments
First-line treatment for AE involves corticosteroids combined with either IVIg or plasma-pheresis22,44:
Methylprednisolone 1 g daily for 5 days, followed by oral prednisone 1 mg/kg (maximum dose 60–80 mg daily, with a prolonged taper over 3–6 months)
IVIg 400 mg/kg/day for 5 days
Plasmapheresis, with 5 exchanges over 7 to 8 days.
Second-line treatments
Second-line treatments can be given as monotherapy or in combination for refractory disease activity 1 to 2 weeks from completion of first-line treatment22:
Rituximab 1,000 mg IV, with a repeat dose in 2 weeks, or 375 mg/m2 weekly IV infusion for 4 weeks
Cyclophosphamide 750 mg/m2 IV monthly for 3 to 6 months.
Maintenance treatments45
Rituximab, repeat every 6 months, same dosing schedule as second-line therapy
IVIg 0.4 g/kg every 2 to 4 weeks
Mycophenolate mofetil 500 to 3,000 mg/day
Azathioprine 1 to 3 mg/kg/day
Cyclophosphamide 1 to 2 mg/kg/day orally.
HOW DO I MONITOR RESPONSE TO TREATMENT IN AE?
There are no validated biological markers to assess treatment response in AE. Although some studies have suggested that early reduction in titers can correlate with better clinical outcomes, antibodies can remain positive even in patients with good outcomes.46 Further, the change in antibody titers does not consistently correlate with risk of relapse.47 Therefore, serum or CSF antibody titers in isolation are not reliable for monitoring treatment response in AE.
Imaging, when abnormal, can be repeated to look for improvements over time. Unfortunately, even after appropriate treatment is initiated for AE, brain MRI may show irreversible changes such as generalized or focal atrophy on follow-up.
Monitoring of the treatment response is therefore primarily based on the clinical examination. We urge clinicians to use objective measures to determine the true efficacy of a given treatment. For example, clinicians can use the Scale for the Assessment and Rating of Ataxia (SARA),48 Symbol Digit Modalities Test, or the Montreal Cognitive Assessment (MoCA)49 for reliable scores to measure the success of a trial and the utility of long-term treatments. Formal neuropsychological testing is also a valuable tool to document the extent of cognitive damage and to evaluate immunotherapy response.
WHAT ONCOLOGIC EVALUATION IS APPROPRIATE FOR PATIENTS WITH AE?
Paraneoplastic AE can occur in association with an underlying malignancy. In particular, small-cell lung cancer, non-small-cell lung cancer, and neuroblastoma trigger the production of paraneoplastic autoantibodies.50 The neurologic sequelae can occur prior to detection of the cancer allowing for early discovery and oncologic treatment.
Though treating the cancer is the main goal in these patients, they may still require short-term or long-term immunotherapy for the paraneoplastic disorder. Immunotherapies that may have dual benefit to treat the cancer and autoimmune disorder, such as cyclophosphamide, can be considered.
All treatment decisions must be made in coordination with the treating oncologist.6 Additionally, patients may have permanent neurologic deficits with relatively little hope for recovery.
The type and frequency of screening for malignancy depends on the specific antibody syndrome.50 If a patient is diagnosed with an autoantibody commonly associated with paraneoplastic syndromes, frequent monitoring is recommended. In those cases, we typically order surveillance testing every 6 to 12 months for at least 3 to 5 years. If the patient has an antibody with a low likelihood of an underlying neoplasm, we consider reevaluating once a year for 3 years after the index diagnosis.50
Based on the 2011 European Federation of the Neurological Societies guidelines, we will utilize either whole-body FDG-PET or CT of the chest, abdomen, and pelvis as a screening measure for certain AE patients, with clinical consideration of the known cancer associations, cancer risk factors, or family history of cancer.50
In particular, the nature of the antibody determines the risk and type of an underlying malignancy and therefore the investigation. For example, anti-Yo autoantibody has a greater than 90% association with breast or ovarian malignancy, so an extensive evaluation should be pursued to identify the underlying neoplasm.50
In addition, sex-specific tests such as pelvic ultrasonography, mammography, and testicular ultrasonography should be considered if the initial evaluation is unrevealing. Further, all patients should complete routine age- and sex-appropriate screening measures including screening for breast, colorectal, cervical, and prostate cancer, along with lung cancer when applicable, based on US Preventive Services Task Force recommendations.
TAKE-HOME MESSAGE
Our understanding of AE has expanded dramatically over the past few years. Familiarity with different AE syndromes will ensure prompt diagnosis and treatment. In cases that are less clear, a sound diagnostic approach anchored on objective clinical or radiographic findings is important for optimizing outcomes. Clinicians need to stay abreast of developments in this field as the breadth of immune-mediated disorders of the nervous system continues to evolve.
DISCLOSURES
The authors report no relevant financial relationships which, in the context of their contributions, could be perceived as a potential conflict of interest.
- Copyright © 2021 The Cleveland Clinic Foundation. All Rights Reserved.
REFERENCES
In this issue




{kind=link}
{kind=link}
Jump to section
- Article
- ABSTRACT
- GENERAL FEATURES OF AUTOIMMUNE ENCEPHALITIS
- HOW IS AUTOIMMUNE ENCEPHALITIS CLASSIFIED?
- WHEN SHOULD I CONSIDER THE DIAGNOSIS?
- WHAT ARE THE COMMON CELL-SURFACE/SYNAPTIC ANTIBODY SYNDROMES IN AUTOIMMUNE ENCEPHALITIS?
- WHAT TESTING SHOULD I CONSIDER FOR AE DIAGNOSIS?
- A COMPREHENSIVE EVALUATION FOR AUTOIMMUNE ENCEPHALITIS
- WHICH AUTOANTIBODY FINDINGS SHOULD BE INTERPRETED CAUTIOUSLY?
- WHAT IS THE INITIAL TREATMENT FOR AUTOIMMUNE ENCEPHALITIS?
- HOW DO I MONITOR RESPONSE TO TREATMENT IN AE?
- WHAT ONCOLOGIC EVALUATION IS APPROPRIATE FOR PATIENTS WITH AE?
- TAKE-HOME MESSAGE
- DISCLOSURES
- REFERENCES
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.