


ABSTRACT
A consequence of chronic and end-stage kidney disease is a higher risk of calcium deposition in sites other than the bones. The authors of this review outline current understanding of the pathogenesis, presentation, diagnosis, and treatment of this group of disorders.
Extraosseous calcification is a broad term that encompasses vascular calcification, soft tissue calcification, and calciphylaxis, all of which are seen in patients with end-stage kidney disease.
The pathogenesis of extraosseous calcification is an active process involving a complex interplay of abnormal electrolyte levels, cell differentiation, and dysregulation of many biochemical pathways.
Vascular calcification is predominantly diagnosed incidentally, while soft tissue calcification and calciphylaxis are diagnosed on the basis of radiographic and clinical presentation, sometimes requiring biopsy.
Management is based on low-quality evidence and includes maintaining a neutral calcium balance, correcting hyperphosphatemia, and controlling comorbidities. Surgical and other nonmedical therapies may help somewhat in managing calciphylaxis and soft tissue manifestations.
Chronic kidney disease, defined as an estimated glomerular filtration rate (eGFR) less than 60 mL/min/1.73 m2 or structural kidney damage sustained over 3 months, is increasing in prevalence worldwide. It is estimated to affect between 2% and 17% of all adults, and the United States is at the high end of this prevalence range.1
As chronic kidney disease progresses, it leads to higher rates of bone mineral disease, a systemic disorder involving the following:
Abnormalities in serum calcium, phosphate, parathyroid hormone (PTH), and vitamin D levels
Disorders of bone metabolism (renal osteodystrophy)
Calcium deposition in both vascular and soft tissues.2
Patients with end-stage kidney disease are at high risk of complications from disorders of bone metabolism, which are strongly associated with increased rates of cardiovascular and all-cause mortality.3–6
NAMES AND PRESENTATIONS
Extraosseous tissue calcification can involve both vascular tissues (arteries and heart valves) and soft tissues. A variety of terms have been used to describe it, based on the location and the type of tissue involved (Table 1), but subclassifying it precisely and studying its prevalence are challenging because its presentation is heterogeneous.
Common terms used to describe calcification
REGULATION OF CALCIUM AND PHOSPHATE
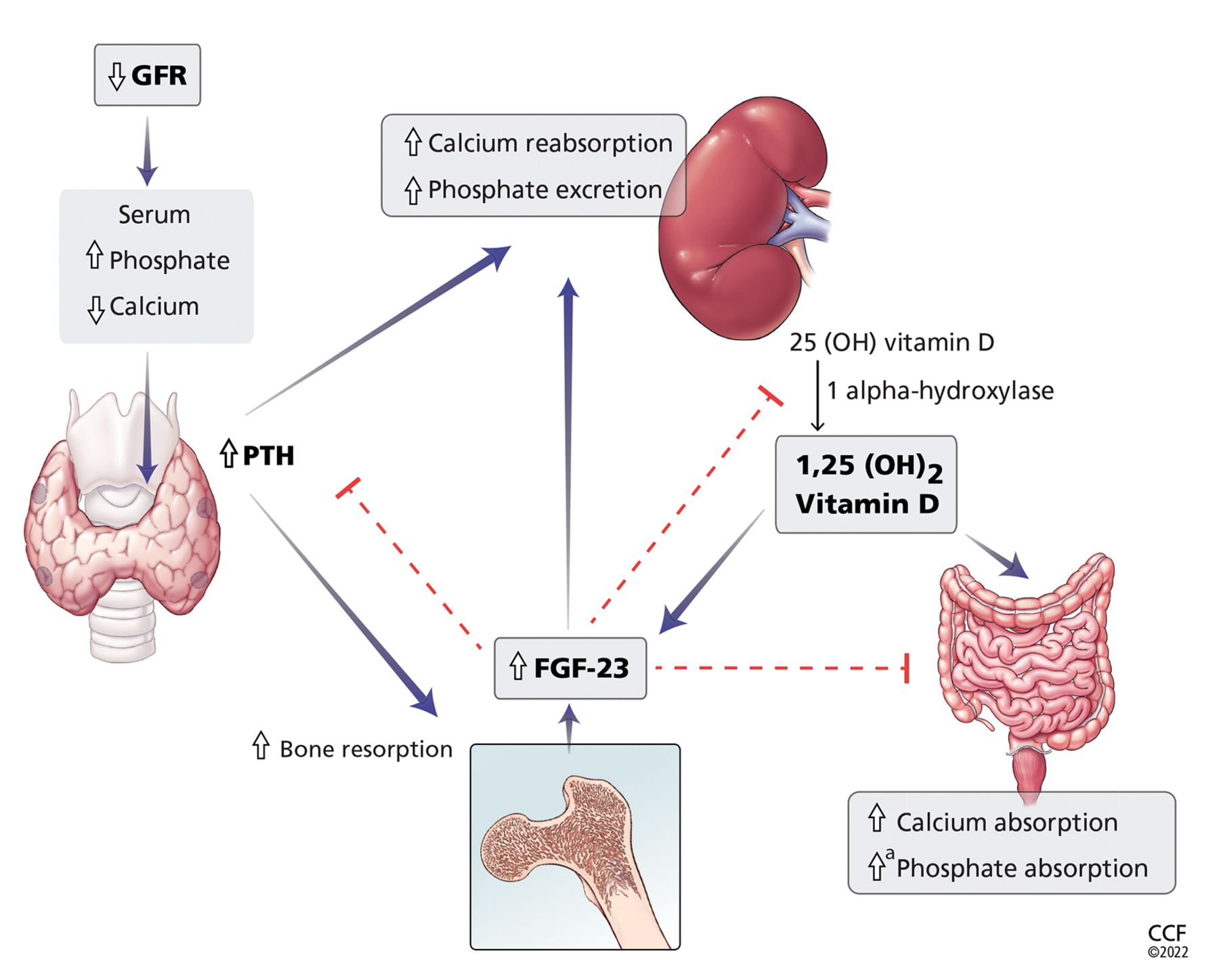
Serum calcium and phosphate levels are kept under tight control by regulatory hormones released by various organs, with complex feedback mechanisms (Figure 1).

Calcium and phosphate metabolism in chronic kidney disease. Decreased glomerular filtration rate (GFR) leads to changes in serum calcium and phosphate, triggering release of parathyroid hormone (PTH) from the parathyroid glands and fibroblast growth factor 23 (FGF-23) from osteoblasts and osteocytes. These hormones have complex downstream effects on the kidney, gut, and bone, both from direct effects on the tissue and from indirect effects through modulation of enzyme activity in vitamin D conversion.
aMinimally increased.
25(OH) vitamin D = 25-hydroxycholecalciferol; 1,25(OH)2 vitamin D = 1,25-dihydroxycholecalciferol
Interestingly, both calcium and phosphate are regulated by the same hormone, ie, PTH.7,8 When serum calcium levels are low and serum phosphate levels are high, the parathyroid glands release more PTH, which acts in several organs to raise the calcium and, on the whole, to lower the phosphate levels.
In the kidney, PTH directly increases calcium reabsorption in the distal tubule and loop of Henle and increases phosphate excretion by inhibiting its reabsorption in the proximal tubule.9,10 Also in the kidney, PTH upregulates production of 1 alphahydroxylase, leading to increased conversion of active vitamin D (1,25-dihydroxycholecalciferol) from its precursor, 25-hydroxycholecalciferol. In turn, in the intestine, active vitamin D increases the absorption of calcium and to a lesser degree phosphate, and in the bone, it has direct actions on both osteoblasts and osteocytes, promoting maturation, expression of skeletal hormones such as fibroblast growth factor 23 (FGF-23), and proper mineralization.11,12
FGF-23 is an important skeletal hormone that lowers phosphate levels by promoting its wasting (ie, suppressing its reabsorption) in the kidney, suppressing its absorption in the intestine, and, in a negative feedback loop, lowering both PTH and 1,25-dihydroxycholecalciferol production.13 Klotho, a protein that has multiple effects in many tissues, facilitates binding of FGF-23 to FGF receptor 1 in the kidney, leading to fewer phosphate receptors in the proximal convoluted tubules, more phosphate excreted in the urine, and lower serum phosphate levels.14 The net effect of these interactions is homeostatic balance in serum calcium and phosphate levels.
CALCIUM-PHOSPHATE AXIS DERANGEMENTS
In chronic kidney disease, nephrons are progressively lost. Among the ill effects is a higher phosphate level, which in turn upregulates production of FGF-23 by the osteocytes and osteoblasts and leads to bone mineral disease (Figure 1). Bone mineral disease can begin early in the course of chronic kidney disease,15 when the eGFR may still be as high as 69 mL/min/1.73 m2. Meanwhile, klotho production is downregulated, so that less FGF-23 binds to its receptor in the kidney,16,17 less 1 alpha-hydroxylase and active vitamin D are produced, and more phosphate is reabsorbed in the proximal convoluted tubule.18,19
As chronic kidney disease progresses to its end stage, FGF-23 levels keep getting higher, and the elevation is accompanied by other calcium-phosphate axis derangements such as excess PTH release, decreased 1,25-dihydroxycholecalciferol, and increased sclerostin (an inhibitor of bone formation).20,21 Together, these derangements lead to the clinical manifestations described below.
Vascular calcification
Vascular calcification is an active process involving de-differentiation of vascular smooth muscle cells. It begins with amorphous development of calcium phosphate nanocrystals in conjunction with other calcium-regulatory proteins in the wall of the artery.22 Deposition of these nanocrystals can begin in the intima of the artery near sites of cholesterol buildup, either progressing into the media or beginning in the media itself, the latter of which is most specific to kidney disease.
In end-stage kidney disease, progression of vascular calcification occurs earlier than in normal aging and is likely driven by hyperphosphatemia, a positive calcium balance, inflammation, and dysregulation between pro-calcification and anticalcification regulatory factors. An in-depth discussion of the pathogenesis of vascular calcification is beyond the scope of this paper and can be found elsewhere.23
Soft tissue calcification
Soft tissue calcification is fairly common in chronic and end-stage kidney disease, but only a small number of patients develop tumoral calcinosis, characterized by massive calcium phosphate deposition in periarticular locations predisposed to microtrauma.
Tumoral calcinosis is well described in families, with autosomal-recessive inheritance stemming from a number of genes, including loss-of-function mutation of FGF23 and missense mutation of alpha-Klotho, contributing to the hyperphosphatemia.24 Hyperphosphatemia is likely a necessary contributor to these familial forms of tumoral calcinosis, but it may also explain their presence in chronic and end-stage kidney disease, stemming from local tissue production or from exogenous phosphate retention.25,26
PRESENTATION AND DIAGNOSIS
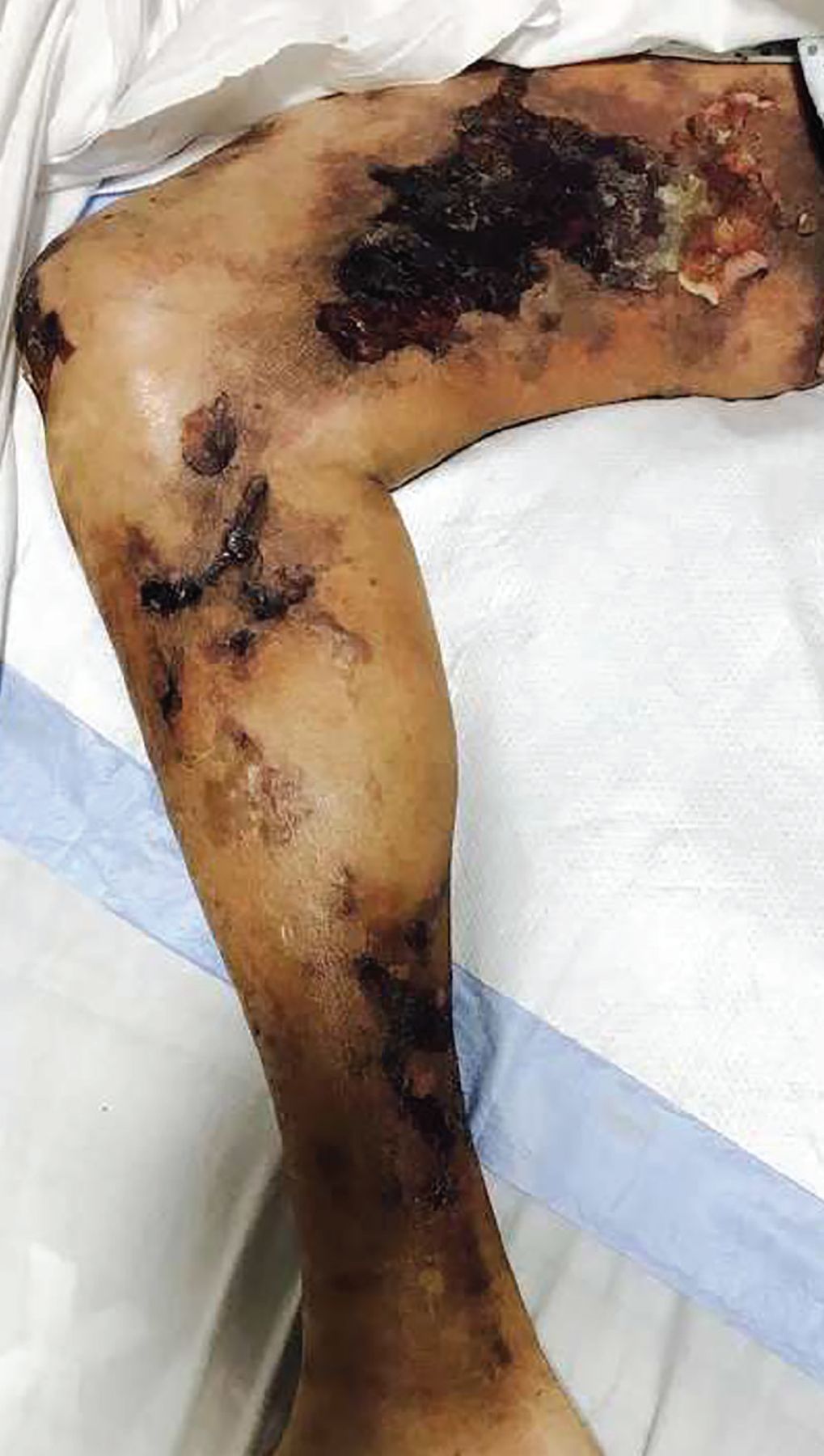
Calciphylaxis is intensely painful, unlike other presentations (Figure 2).27 It is most commonly seen in adipose-dense tissues but can develop centrally and in appendicular areas, including the genital regions. Skin lesions can vary from induration to ulceration with eschar formation.28 Its diagnosis is predominantly clinical. A skin biopsy to the depth of the subcutaneous tissue can aid diagnosis but poses significant procedural risks that include pain intensification, poor healing, and secondary infection.29

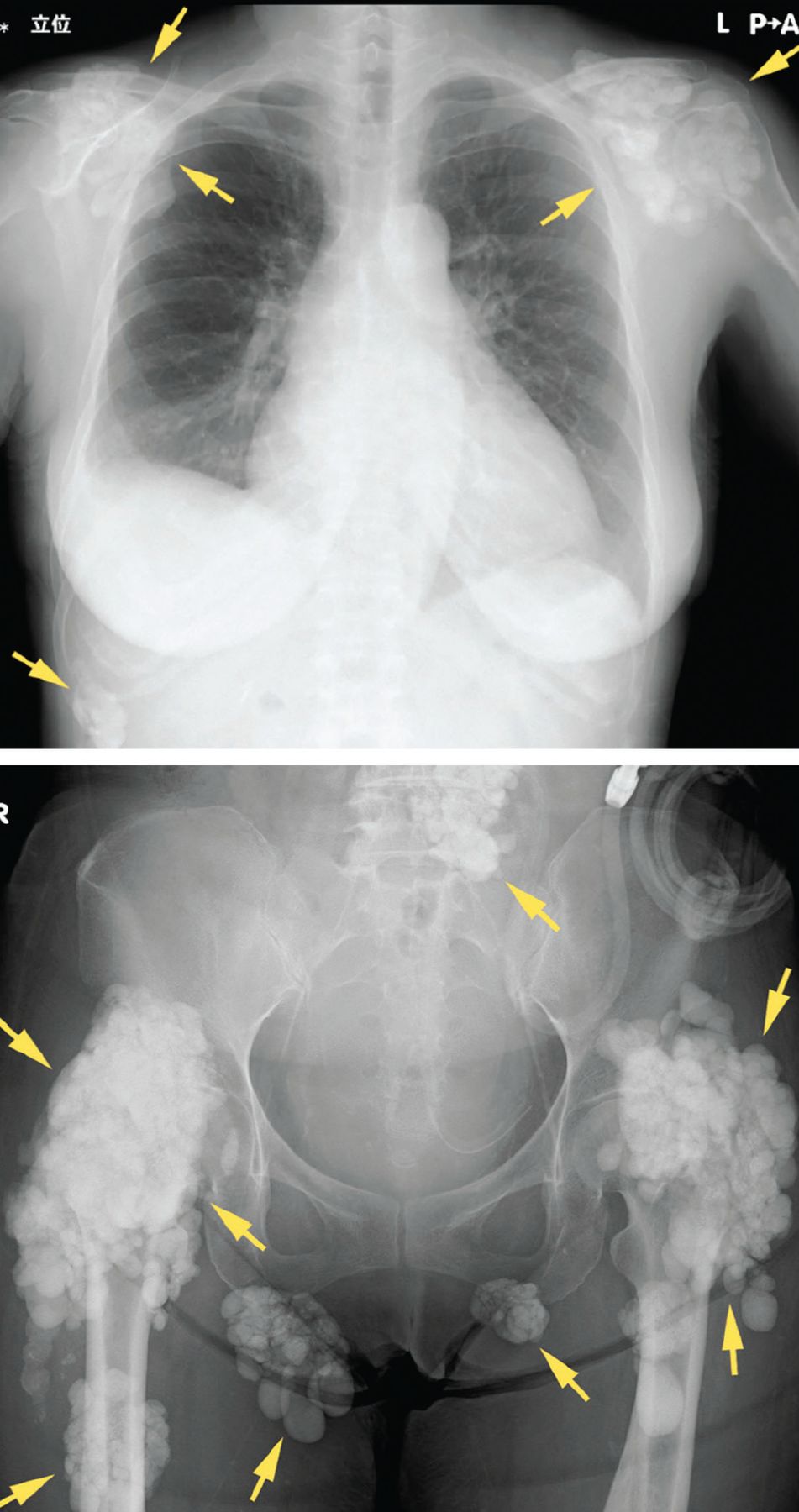
Soft tissue calcifications, in contrast, are usually painless, unless radicular symptoms develop from mass effect. Instead, there is typically a decrease in range of motion of the affected joints,30 of which (in descending order of frequency) the hip, elbow, shoulder, foot, and wrist are most commonly affected (Figure 3).31 Soft tissue calcifications tend to be formally diagnosed based on the location of the calcium deposition, in addition to morphologic descriptions to rule out cancer mimickers.

Radiography shows calcified masses (arrows) in a 47-year-old woman with tumoral calcinosis.
From reference 30.
Vascular calcification. Traditional risk factors that predict atherosclerotic calcification do not fully explain the high prevalence of vascular calcification in patients with chronic and end-stage kidney disease. Additional potentially modifiable risk factors related to kidney disease or its treatment have been shown to accelerate calcification (Table 2).32
Risk factors associated with vascular calcification
MEDICAL THERAPY
Most of the research has focused on therapies directed at vascular calcification, given its clinical implications with cardiovascular disease in end-stage kidney disease.
Dietary phosphate restriction, phosphate binders
Given the central role of elevated phosphate and FGF-23 in the pathogenesis of extraosseous calcification, controlling serum phosphate levels, first through dietary phosphate restriction and then with intestinal phosphate binders, is a logical and low-cost management choice in preventing vascular calcification.
The most commonly used phosphate intestinal binders are calcium-based (eg, calcium carbonate, calcium acetate) and are used extensively in patients with chronic and end-stage kidney disease for many indications. However, earlier studies demonstrated a relationship between higher calcium intake and higher rates of vascular calcification,33 and subsequent studies called attention to this association, leading to recommendations for using non–calcium-based intestinal phosphate binders to restore normal phosphate levels while limiting calcium intake to maintain normal serum calcium.34,35
A number of randomized trials over the last 20 years have attempted to settle the debate on calcium-based vs non–calcium-based phosphate binders and cardiovascular disease, many of them using vascular calcification as a surrogate end point.
The IMPROVE-CKD trial36 (Impact of Phosphate Reduction on Vascular End-points in Chronic Kidney Disease) tested lanthanum use in patients with advanced chronic kidney disease (eGFR < 30 mL/min/1.73 m2) and evaluated changes in aortic calcification and arterial stiffness. It did not find statistically significant differences with lanthanum compared with placebo. Of note, the trial was limited by recruitment, including patients with normal phosphate levels and excluding those with end-stage kidney disease.36
The Treat-to-Goal study37 in patients with end-stage kidney disease on hemodialysis found less coronary artery and aortic calcification and a lower incidence of hypercalcemia in those randomized to sevelamer compared with calcium acetate. These results may correlate with improved all-cause survival rates in patients newly started on hemodialysis, despite lower rates of normophosphatemia when sevelamer is used.15,38 Subsequent studies comparing lanthanum carbonate with calcium carbonate in patients newly starting on hemodialysis did not find statistically significant differences in calcification scores in heart valves.39
The LANDMARK trial40 (Outcome Study of Lanthanum Carbonate Compared With Calcium Carbonate on Cardiovascular Mortality and Morbidity in Patients With Chronic Kidney Disease on Hemodialysis), published in 2021, looked at patients with end-stage kidney disease in Japan who had risk factors for vascular calcification who were randomized to receive lanthanum or calcium carbonate. It did not find any statistically significant differences in rates of all-cause mortality or cardiovascular events between the two groups, though the event rates were low. Further, compared with the United States, Japan has lower dietary calcium intake, higher use of arteriovenous fistulas for dialysis access, and different cardiovascular screening practices, which could limit wide applicability of the results.40
In sum, data conflict regarding whether non–calcium-based intestinal phosphate binders are superior to calcium-based binders in preventing vascular calcification and cardiovascular events.
Bone antiresorptive agents
Pyrophosphates (bisphosphonates), the most commonly used class of drugs for preventing bone resorption, inhibit the activity of osteoclasts, and some of these drugs also induce apoptosis. Bisphosphonates are either retained in the bone or cleared by the kidney.
Robust data exist for using this drug class in bone disorders in patients in the early stages of chronic kidney disease (eGFR > 35 mL/min/1.73 m2), but data are significantly limited in those with stage 4 or 5 chronic kidney disease or end-stage kidney disease, and there are theoretical safety concerns.41 Bisphosphonates are less frequently prescribed in these latter populations, possibly due to concerns about toxicity, as these drugs are excreted by the kidney.42 Reports of worsening kidney disease or kidney injury exist for most drugs in the bisphosphonate class, but larger observational trials have found oral bisphosphonates to be reasonably safe in advanced chronic kidney disease, though bisphosphonate users had a 14% higher risk of progression of chronic kidney disease.43
Zoledronic acid, a potent intravenous formulation, should be avoided if the eGFR is less than 30 mL/min/1.73 m2, in view of stronger associations with direct tubular injury, acute kidney injury, and worsened eGFR.44,45 Pamidronate is generally the preferred intravenous formulation for patients with advanced chronic kidney disease, usually given at a lower dose or infused over a longer time. Rarely, collapsing focal segmental glomerulosclerosis can occur.44,45
Bisphosphonates have been shown to reduce both overall vascular calcification and all-cause mortality in certain groups (eg, patients with osteoporosis or cancer), but not the rate of cardiovascular events.46 Etidronate, a first-generation bisphosphonate now discontinued due to high rates of osteomalacia, was used to treat soft tissue calcifications.47–49 Etidronate also reduced vascular calcification in rat models of chronic kidney disease, while human studies showed reduced coronary artery calcification in patients with advanced chronic kidney disease and end-stage kidney disease.49–51 Newer bisphosphonates have limited data on their effects on vascular calcification in end-stage kidney disease, with one study of alendronate showing no improvement in coronary artery calcification score.52
Denosumab, a RANK ligand inhibitor (RANK stands for receptor activator of nuclear factor kappa B) that prevents osteoclast maturation, has not been studied in soft tissue calcification. Small pilot studies have looked at denosumab’s effects on vascular calcification in humans and have suggested it may slow coronary artery calcification, but this has been challenged in other studies.52,53 More studies are needed to determine the clinical significance of these findings. We are not aware of any studies that have looked at denosumab in soft tissue calcification or calciphylaxis.
Teriparatide is a synthetic formulation of PTH. The only evidence for using it to treat tumoral calcinosis comes from case reports, and no major studies have looked at using it in end-stage kidney disease to prevent vascular calcification.54
Calcimimetics
Calcimimetics are drugs that bind allosterically to the calcium-sensing receptor on parathyroid cells to suppress PTH release for a given serum calcium level.
Cinacalcet, the most common drug in this class, has been studied extensively in secondary hyperparathyroidism in 2 trials, the EVOLVE55 (Evaluation of Cinacalcet Hydrochloride Therapy to Lower Cardiovascular Events) and ADVANCE56 (A Randomized Study to Evaluate the Effects of Cinacalcet plus Low-Dose Vitamin D on Vascular Calcification in Subjects With Chronic Kidney Disease Receiving Heemodialysis). It did not show improvement in aortic calcification or reduction in cardiovascular outcomes or all-cause mortality despite improvements in serum PTH levels.55,56 In contrast, a more recent meta-analysis of cinacalcet use in end-stage kidney disease did find a benefit in terms of lower rates of all-cause mortality and cardiovascular mortality.57 Other calcimimetics have been studied only in animal models, and thus their clinical effect in humans is undetermined.
Sodium thiosulfate
Sodium thiosulfate is an older medication with antioxidant properties that has been used off-label for years in calcium disorders including vascular calcification and calciphylaxis. It was recently systematically reviewed in treating calciphylaxis, with conflicting results.58,59 More recently, a randomized clinical trial60 showed reduction of iliac artery calcification and arterial stiffness with sodium thiosulfate compared with placebo in calciphylaxis. Ongoing prospective and randomized trials will hopefully provide clarity of the benefit of sodium thiosulfate in vascular calcification and calciphylaxis. In a small case series, the drug has shown improvement in symptom burden in soft tissue calcification of the shoulder and hip, with partial size regression.61
Vitamin K
Vitamin K is an essential cofactor for carboxylation of numerous proteins, including some that inhibit vascular calcification, such as matrix G1a protein.62 Evidence that lack of vitamin K may be involved in vascular calcification includes a high prevalence of vitamin K deficiency in this population and improvement in carboxylation surrogate markers with supplementation.63,64
Warfarin, a vitamin K antagonist, accelerates medial arterial calcification, particularly in end-stage kidney disease.65 Furthermore, warfarin has been identified observationally as a risk factor for calciphylaxis, and low levels of carboxylation of matrix G1a protein are associated with calciphylaxis in end-stage kidney disease.66 The suspected mechanism by which warfarin may contribute to calciphylaxis is by inhibiting vitamin K-dependent carboxylation of matrix G1a protein, a mineral-binding extracellular matrix protein that prevents calcium deposition in arteries.
Several phase 3 trials are being conducted to determine the benefit of vitamin K supplementation in vascular calcification and calciphylaxis, though a recent trial in patients with stage 4 chronic kidney disease67 did not show improvement in vascular stiffness with vitamin K supplementation. There are no current studies looking at tumoral calcinosis and vitamin K supplementation.
Novel therapies, nonmedical management
SNF472, a myoinositol hexaphosphate that inhibits hydroxyapatite growth, has shown promise in early clinical trials in reduction of coronary artery calcium volume, while tissue-nonspecific alkaline phosphatase inhibitors are in earlier stages of development.68,69
Magnesium and vitamin D supplementation in chronic and end-stage kidney disease has had varying degrees of success in preventing vascular calcification, though more studies are needed to confirm its clinical utility.70,71 With particular relevance to soft tissue calcification, surgical debridement and hyperbaric oxygen therapies hold significant promise as adjunctive therapies to the aforementioned medical therapies.72–74
DISCLOSURES
Dr. Fatica has disclosed working as an advisor or review panel participant for Natera Inc and REATA Pharmaceuticals. The other authors report no relevant financial relationships which, in the context of their contributions, could be perceived as a potential conflict of interest.
- Copyright © 2022 The Cleveland Clinic Foundation. All Rights Reserved.
REFERENCES
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.