


ABSTRACT
Acute cardiac injury, defined as an elevated high-sensitivity troponin I or troponin T upon admission or during hospitalization, is common in patients with COVID-19, occurring in 10% to 35% of patients depending on the assay used and the population studied. Even though the mechanisms of SARS-CoV-2 myocardial injury are not well defined, type 1 myocardial infarction and fulminant myocarditis are rare. Often, acute cardiac injury occurs in patients with elevated inflammatory markers, and both are associated with worse outcomes. However, the extent to which treatments should differ for patients with acute cardiac injury, heightened systemic inflammation, or both, is unknown.
The mechanisms of acute cardiac injury in COVID-19 are still being defined but include oxygen supply-demand imbalance, microvascular and endothelial dysfunction, and micro- and macrothrombosis. In some patients, these manifestations may be driven by an inappropriate inflammatory response.
Like other patients, COVID-19 patients with ischemic ST-segment elevation need emergency reperfusion therapy.
Patients with elevated troponin and elevated inflammatory markers may possibly benefit from immunosuppressive therapy, although further studies are needed.
Initial case-fatality rates in coronavirus disease 2019 (COVID-19) have ranged from 2.3% to 7.3%,1,2 and given the burden of disease, the devastation is singularly alarming and unprecedented. Even though the predominant manifestations are respiratory, concomitant cardiovascular complications result in substantial morbidity and mortality.3
Acute cardiac injury in COVID-19 due to infection with SARS-CoV-2 has been defined primarily as an elevation in serum cardiac markers above the 99th percentile upper reference range, as it was in prior investigations of other viral infections, and the incidence has ranged from approximately 8% to 36%.4–8 Using the broad and inclusive definition of acute cardiac injury as an elevated high-sensitivity troponin I or troponin T upon admission or during hospitalization, the mortality rate has been striking—over 50% in initial reports.5,6
Given this startling signal, amid our ever-changing understanding of this pandemic, the following questions warrant emphasis:
What is the mechanism of SARS-CoV-2–associated myocardial injury?
To what extent are SARS-CoV-2 patients with myocardial injury a distinct population?
What are possible treatment options for myocardial injury associated with SARS-CoV-2 infection?
WHAT IS THE MECHANISM?
With regard to mechanism, the primary question is whether SARS-CoV-2 infection precipitates myocardial infarction with an oxygen supply-demand imbalance, either with or without acute coronary plaque pathology (type 1 and 2 myocardial infarction), or conversely, causes myocardial injury mediated by the virus itself or the cytokine response to it.
Viral infections are well known to lead to adverse cardiovascular events, either by increased metabolic demand in the setting of limited cardiac reserve or by precipitating plaque rupture in the setting of inflammation and a prothrombotic state.9 Of note, influenza vaccination has been shown to reduce hospitalizations for cardiac disease.10 In addition, certain viruses (eg, parvovirus B19 and influenza) commonly cause myocarditis.
Although myocardial injury has not been prominent with other coronaviruses, unfortunately, SARS-CoV-2 appears to be behaving differently. Despite an overall case-fatality rate of approximately 10% in symptomatic patients in the previous SARS-CoV outbreak that resulted in severe acute respiratory syndrome (SARS), cardiac complications were anecdotal and limited to case reports or series.11 Similarly, despite an even higher case-fatality rate in Middle East respiratory syndrome due to MERS-CoV, cardiac complications were limited.9,12
In contrast, in an initial report of causes of death in COVID-19, one-third were considered secondary to respiratory failure with myocardial damage, and nearly another tenth were considered secondary to myocardial damage alone.13 Furthermore, perimyocarditis from SARS-CoV-2 infection has been reported in the absence of symptomatic respiratory disease,14 though fulminant myocarditis—generally defined as sudden and severe inflammation of the myocardium resulting in myocyte necrosis, edema, and cardiogenic shock—seems to be a rare presentation with SARS-CoV-2.
Of note, SARS-CoV-2 enters respiratory and cardiac cells via angiotensin-converting enzyme 2 (ACE 2), a membrane-bound protein.3,9 Yet this potential cardiac tropism offers an incomplete explanation for the seemingly disproportionate cardiac manifestations of COVID-19, given that SARS-CoV also uses ACE 2 as a functional receptor.15,16
Alternatively, myocardial injury may be exacerbated by an inappropriate activation of type 1 T-helper cells and cell-mediated immunity with associated cytokine storm.3 A recent autopsy study17 is consistent with this hypothesis. Among 39 patients, SARS-CoV-2 cardiac infection was documented in 61.5%, and patients with higher viral loads had greater expression of proinflammatory genes. However, inflammatory cell infiltrates typical of active myocarditis were not observed.
A DISTINCT POPULATION?
These putative mechanisms of injury are integrally entwined with the second question of whether patients with SARS-CoV-2–associated myocardial injury represent a distinct population. For one, COVID-19 patients with elevated troponins are older and have more cardiovascular comorbidities, such as coronary artery disease, chronic heart failure, hypertension, and diabetes mellitus.5,6,8 These findings support the concept of myocardial oxygen supply-demand mismatch with resultant ischemia in a vulnerable population.
However, in patients who succumb to COVID-19, troponin levels may continue to rise throughout the illness, a pattern distinct from the typical rise and fall after an ischemic insult.6 Moreover, about a third of patients may demonstrate an increase in troponin over time, and these patients have a higher mortality rate.8 Importantly, patients with elevated troponins have higher levels of inflammatory markers such as C-reactive protein (CRP).8 The increases in troponin and CRP appear to parallel each other, and the overall correlation is similar in magnitude to the correlation between troponin and N-terminal probrain natriuretic peptides. These observations, though nascent, suggest that some patients may develop a hyperinflammatory state that perpetuates nonischemic myocardial injury.
Given that elevated troponin is associated with a high mortality rate and that the mechanism of injury could be related to increased systemic inflammation, as more data are emerging, consideration should be given to checking troponin upon admission, with surveillance testing during the initial days of hospitalization. Further considerations in this initial clinical approach include assessing cardiac risk factors and the magnitude of the inflammatory response.
A rise and fall of cardiac markers in the presence of signs and symptoms of myocardial ischemia, such as new ischemic changes on electrocardiography or imaging evidence of regional myocardial dysfunction in a pattern compatible with ischemia, diagnoses a myocardial infarction. The absence of these features defines myocardial injury, and substantial elevations in CRP may point toward cytokine-mediated damage.
Traditionally, cardiac imaging would feature prominently in the distinction between acute myocardial infarction and injury. Given limited resources and the need to minimize exposure to COVID-19 patients, this decision will be individualized, though it will involve selective use of focused echocardiography.
In patients who have convalesced from COVID-19, studies have shown that myocardial damage and inflammation may be evident in a majority of patients when assessed with cardiac magnetic resonance imaging.18,19 However, the cross-sectional design of these studies precludes any assessment of causality, and the clinical implications are unclear. Therefore, in the absence of another indication, cardiac magnetic resonance imaging is currently not clinically recommended in asymptomatic patients who have recovered from COVID-19.
Of note, the overlap between acute myocardial infarction, myocardial injury, and heightened systemic inflammation continues to be defined, though these considerations do aid in risk stratification. An elderly patient with coronary artery disease, diabetes mellitus, and elevations in troponin and CRP will have among the poorest prognoses. However, even in the absence of these risk factors, a patient with elevated troponin and inflammatory markers is at increased risk.8 In evaluating patients with an elevated troponin, we are well accustomed to risk stratification according to cardiovascular comorbidities, but with COVID-19, we should also risk-stratify based on the degree of heightened inflammation.
WHAT ARE THE POSSIBLE TREATMENTS?
Finally, the consideration of treatment options in a patient with a positive troponin test is informed by the presumed answers to the first 2 questions. Specifically, does the mechanism of injury seem more likely related to myocardial infarction with oxygen supply-demand mismatch, or to direct myocardial injury? And is this a patient with underlying cardiac conditions, increased systemic inflammation, or both?
Treatment strategies for type 1 myocardial infarction are well delineated, and treatment of type 2 myocardial infarction includes addressing the underlying cause and providing therapies to improve the myocardial oxygen supply-demand mismatch, especially in the setting of known fixed coronary stenosis. Importantly, therapies such as beta-blockers and vasodilators must be used judiciously to avoid precipitating decompensated heart failure or shock. In this setting, revascularization is rarely indicated, and the benefit of antiplatelet and anticoagulant therapy is unknown.
With COVID-19, treatment is supportive, and directed therapies are urgently required. Remdesivir has been shown to shorten the time to recovery, and in hospitalized patients who are hypoxic, dexamethasone improves survival.20 In severe disease, the success of dexamethasone suggests that morbidity and mortality may be driven by heightened inflammation.21,22 This increased inflammation may progress to secondary hemophagocytic lymphohistiocystosis and resultant fatal hypercytokinemia with multiorgan failure. In this inflammatory state, further immunosuppression may improve outcomes.
In cardiac disease, promising immune treatments target autoinflammation, a process driven by endogenous danger signals and perpetuated by inflammasome-induced cytokine production. Such therapies have demonstrated efficacy and include colchicine, rilonacept, and anakinra in pericarditis and colchicine and canakinumab in atherosclerotic disease.23–27 Colchicine inhibits tubulin polymerization and inflammasome activity, whereas anakinra, rilonacept, and canakinumab inhibit interleukin 1, a cytokine that is central to the inappropriate innate immune response. In open-label case-control studies, anakinra28 and canakinumab29 have shown promise in treating severe COVID-19 pneumonia, and a small randomized trial suggested a potential benefit of colchicine.30 Accordingly, larger randomized studies with these therapies are currently enrolling patients.
FEW ANSWERS, MANY QUESTIONS
For troponin-positive COVID-19 patients, we currently have few answers and many questions. With COVID-19, typical acute coronary syndromes and classic myocarditis occur rarely. The mechanisms of acute cardiac injury are still being defined, but include oxygen supply-demand imbalance, microvascular and endothelial dysfunction, as well as micro-and macrothrombosis. In some patients, these manifestations may be driven by an inappropriate inflammatory response.
In general, for the practicing clinician, we can consider 3 broad categories of patients with COVID-19 and abnormal troponins:
Patients with ischemic ST elevation, who need emergency reperfusion therapy
Patients with troponin elevation without systemic heightened inflammation, who need supportive care
Patients with elevated troponin and inflammatory markers, who may possibly benefit from immunosuppressive therapy, although further studies are needed (Figure 1).
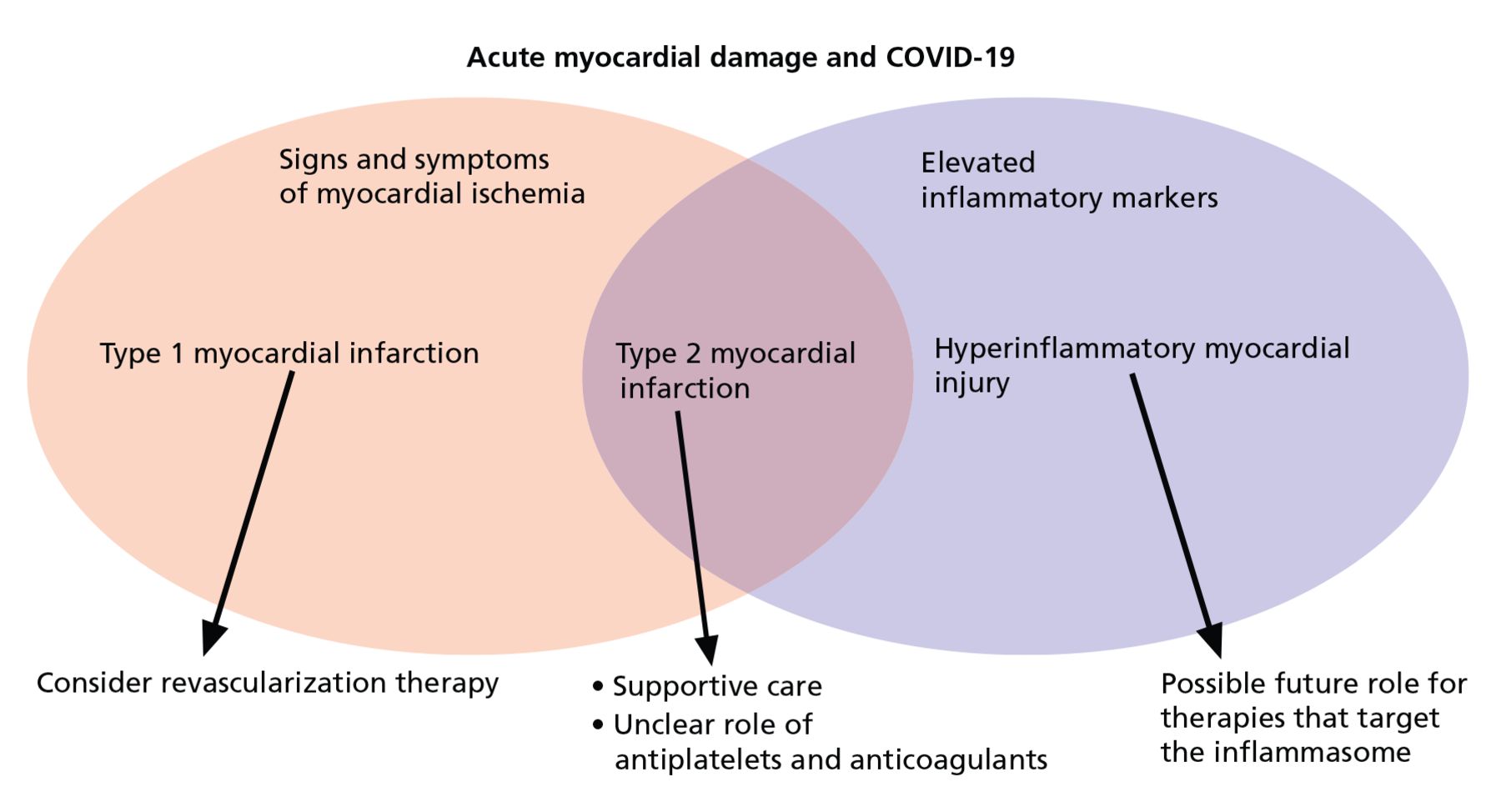
Three broad causes of acute cardiac injury.
- Copyright © 2020 The Cleveland Clinic Foundation. All Rights Reserved.
REFERENCES
In this issue




{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.