


ABSTRACT
Noninfectious aortitis is occasionally detected incidentally, either on imaging or on histopathologic review after open thoracic aortic surgery. It can present as a clinically asymptomatic, seemingly focal lesion, as diffuse inflammation throughout several aortic segments but sparing the branch vessels, or as a manifestation of a widespread systemic condition. Treatment differs based on etiology, so once identified, all patients with aortitis need a thorough evaluation, laboratory tests, complete large-vessel imaging, and a referral to a vasculitis expert. All patients with aortitis are at high risk of future vascular complications and should be followed with serial clinical evaluations and imaging.
Noninfectious thoracic aortitis may be detected radiographically or on histopathologic review after open thoracic aortic surgery.
Aortitis may be a manifestation of a widespread systemic illness, or a form of single-organ vasculitis, termed isolated aortitis.
All patients with aortitis need a complete workup and referral to a vasculitis expert.
Immunosuppression decisions are complex, influenced by the presence or absence of an underlying systemic condition and suspicion of persistent vasculitis, and therefore must be made on an individual basis.
Noninfectious aortitis is occasionally detected incidentally, either on imaging or on histopathologic review after open thoracic aortic surgery. It can be a manifestation of a heterogeneous group of diseases, or in some cases, exist in isolation. Aortitis is an important finding that requires timely management because it can lead to poor outcomes such as aneurysm formation, dissection, or need for vascular surgery.
See related editorial, page 635
Below, we review how aortitis is detected, its many possible causes, and the workup and treatment of patients who are found to have it.
THE AORTA: NOT A SIMPLE TUBE
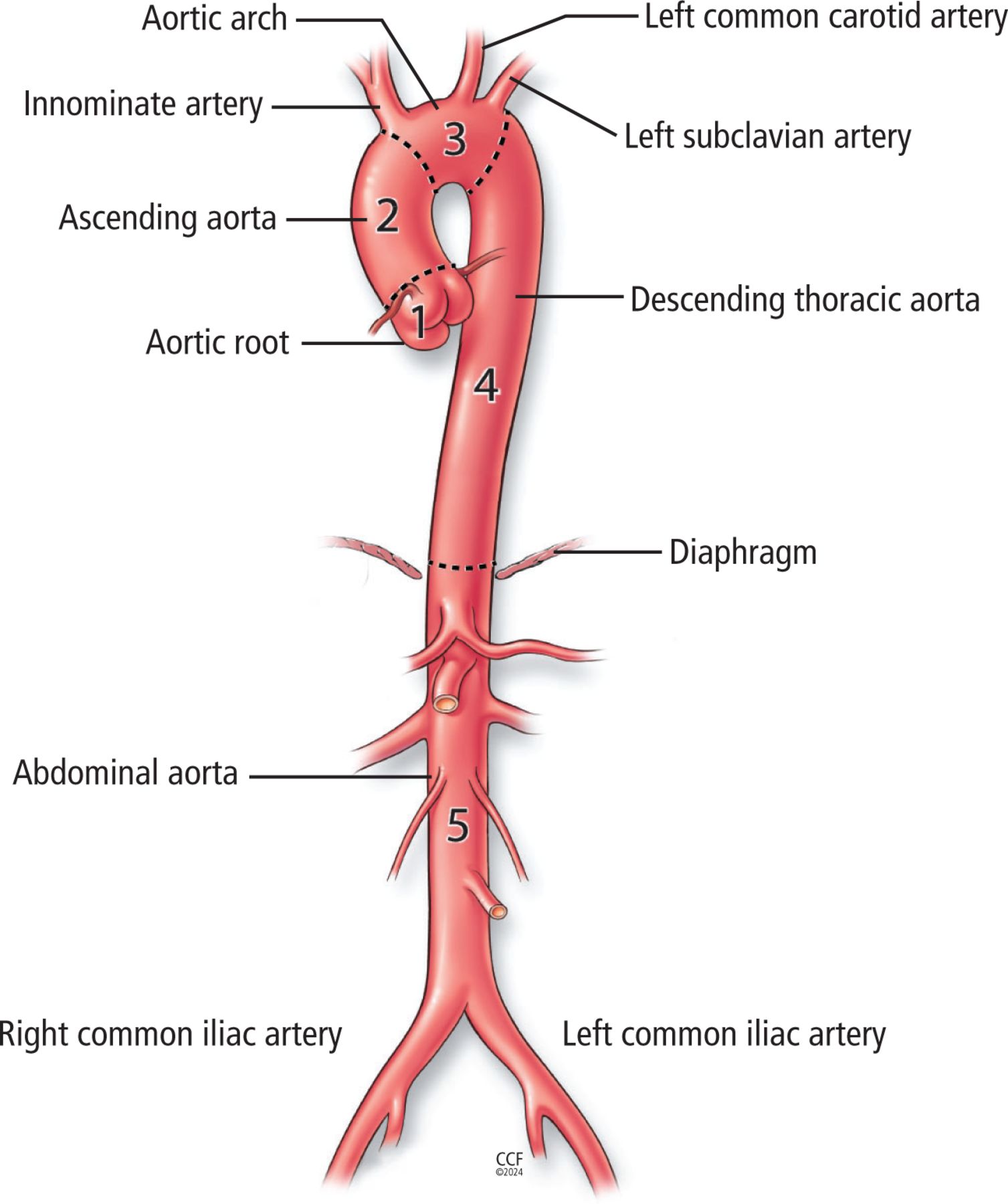
The aorta, the largest blood vessel in the body, is divided into 5 sections: the aortic root, ascending aorta, aortic arch, descending thoracic aorta, and abdominal aorta (Figure 1).1 Although contiguous, aortic segments differ from one another in many important ways, including gene expression; medial wall thickness; number of smooth muscle cells, elastic fibers, and vasa vasorum; and, importantly, susceptibility to disease.2,3 While atherosclerosis most commonly affects the abdominal aorta, noninfectious aortitis preferentially targets the thoracic segments.3,4

Anatomy of the aorta. The 5 segments of the aorta are the (1) aortic root (from the aortic valve through the sinotubular junction), (2) ascending aorta (from the sinotubular junction to the innominate artery), (3) aortic arch (from the innominate through the left subclavian artery), (4) descending thoracic aorta (left subclavian artery to the diaphragm), and (5) abdominal aorta (diaphragm to the iliac bifurcation).
DEFINING AORTITIS: TISSUE VS IMAGING
Thoracic aortitis can be defined either histopathologically or radiographically.
Histopathologic findings
Aortitis is occasionally detected postoperatively in specimens sampled during open thoracic aortic repair.4–8 Four patterns of tissue inflammation have been described,9 and can provide important clues to the underlying condition:
Granulomatous giant cell pattern, characterized by epithelioid macrophages with or without multinucleated giant cells
Lymphoplasmacytic pattern: lymphocytes and plasma cells without a granulomatous component (staining for immunoglobulin [Ig] G4–positive plasma cells is recommended)
Mixed inflammatory pattern: many inflammatory cell types without an overt granulomatous pattern
Suppurative pattern: neutrophilic abscesses with necrosis (staining for microorganisms is strongly recommended).
See Table 1,10–12 Table 2,4–8,10–19 and Table 310–12,20–29 for the differential for these 4 patterns.
Infectious causes of aortitis
Primary vasculitic causes of aortitis
Secondary causes of aortitis
Radiographic findings
Using computed tomographic angiography (CTA) or magnetic resonance angiography (MRA), aortitis is typically defined as circumferential thickening (> 2 to 3 mm) of the aortic wall with contrast enhancement (with or without vessel wall edema in the case of MRA) without atherosclerotic plaque.30,31 Berthod et al32 in 2018 reported that the optimal threshold for distinguishing pathologic aortic wall thickness in patients with giant cell arteritis from controls was 2.2 mm.
Aortitis shares some radiographic similarities with aortic intramural hematoma, an aortic emergency characterized by a crescent-shaped thickening of the aortic wall greater than 5 mm caused by slow bleeding and thrombus formation in the media.1 However, the shape on CTA differs (diffuse, circumferential thickening in aortitis vs focal and crescentic thickening in aortic intramural hematoma), as does the clinical presentation. In uncertain cases in which the patient is clinically stable, positron-emission tomography with computed tomography (PET-CT) can be used to distinguish these 2 processes (in aortitis, the thickened aortic wall avidly takes up 18F-fluorodeoxyglucose [FDG], but in aortic intramural hematoma it does not).33
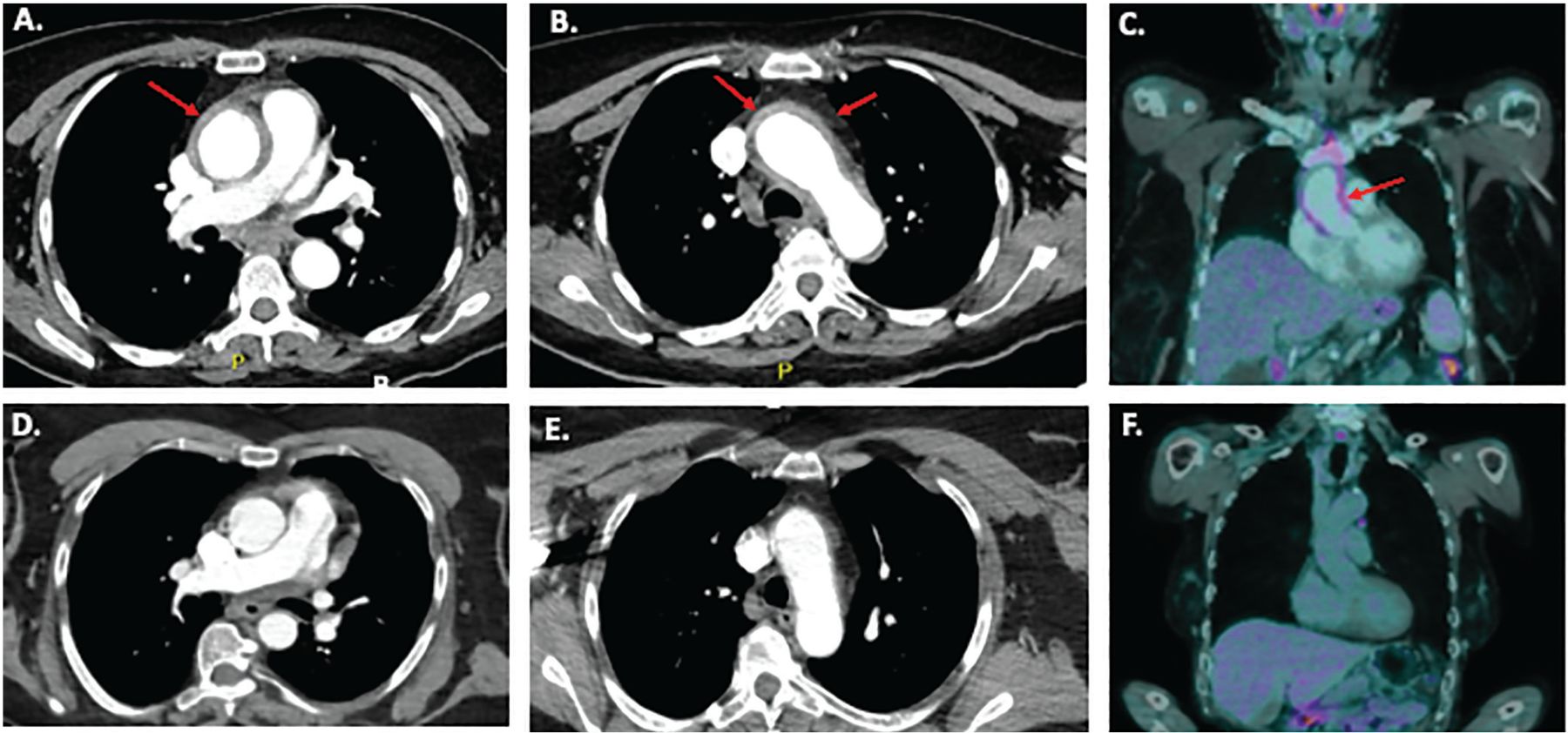
Using PET-CT, FDG uptake in the aortic wall can be visually compared with uptake in the liver: if uptake is similar in both places, it is considered grade 2 (possibly indicative of aortitis), and if higher in the aorta, it is considered grade 3—compatible with aortitis.31 As with CTA or MRA, the pattern of vascular FDG uptake is also important—aortitis produces longer, circumferential FDG-avid lesions, frequently spanning multiple sections of the aorta, while patchy or focal aortic uptake is seen in atherosclerosis.13,31 Figure 2 shows examples of CTA and PET-CT images of thoracic aortitis.

Imaging studies in a 55-year-old female who presented with fever, chest pain, and a C-reactive protein level of 33 mg/L (normal range < 8 mg/L). (A and B) Computed tomography angiography (CTA) of the chest and abdomen showed diffuse circumferential thickening (up to 6 mm) of the wall of the ascending aorta through the aortic arch consistent with aortitis. Red arrows indicate circumferentially thickened and FDG-avid aortic wall in the ascending aorta and aortic arch. There were no symptoms or physical signs of an underlying systemic vasculitis or autoimmune disease. Laboratory tests were normal or negative, including blood cultures, antinuclear antibody, extractable nuclear antigen, anti-double-stranded DNA, complement components 3 and 4, antineutrophil cytoplasmic antibody, serum immunoglobulin G4 level, urinalysis, interferon-gamma release assay for tuberculosis, and serology for hepatitis B and C and syphilis. (C) Positron-emission tomography with computed tomography (PET-CT) showed longitudinal grade 3 18F-fluorodeoxyglucose (FDG) uptake (ie, more than in the liver) in the ascending aorta and arch, consistent with active aortitis. Prednisone 60 mg daily was initiated, symptoms improved, and C-reactive protein level returned to normal. (D and E) CTA images of a normal ascending aorta and arch, shown for comparison. (F) PET-CT images of a normal ascending aorta without pathologic FDG uptake.
Can imaging detect histopathologic aortitis preoperatively?
A single study has directly compared the yield of imaging vs tissue histopathology for the diagnosis of thoracic aortitis. In 16 patients in whom histopathologic evidence of noninfectious aortitis was found following thoracic aneurysm repair, preoperative PET-CT failed to identify aortitis in 5 (31%).34 Even in those in whom PET-CT was positive for aortitis, the preoperative C-reactive protein level was normal in 6 of the 8 in whom this laboratory result was available, indicating that neither imaging nor inflammatory markers are sufficiently sensitive to reliably identify aortitis at the tissue level.
Given that the histopathologic finding of aortitis is rare and preoperative PET-CT imaging does not identify all cases, routine preoperative PET-CT imaging in all patients undergoing thoracic aortic aneurysm repair does not seem warranted.
MANY POSSIBLE CAUSES OF AORTITIS
Aortitis can be a manifestation of numerous conditions, which range in severity from asymptomatic to life-threatening. The differential diagnosis includes infectious causes (Table 1), primary vasculitic diseases (Table 2), and secondary causes (Table 3). Below, we have expanded on a few of the most important noninfectious causes.
Giant cell arteritis
Giant cell arteritis is the most common primary systemic large-vessel vasculitic disease in North America, with an estimated incidence of 15 to 20 per 100,000 per year in people older than 50.14 It classically targets the cranial arteries, including the superficial temporal arteries, facial artery, and posterior ciliary arteries, giving rise to typical symptoms of headache, scalp tenderness, jaw claudication, and vision loss. However, most patients also have concomitant large-vessel involvement.14
In a study of 40 patients with biopsy-proven giant cell arteritis who prospectively underwent imaging, 26 (65%) had radiographic evidence of aortitis at diagnosis.15 Conversely, about one-third of patients with aortitis detected radiographically are subsequently found to have giant cell arteritis.35,36 The thoracic arch and descending thoracic aorta segments are most often involved, followed by the ascending and abdominal aorta.15
Possible red flags for aortitis in patients with giant cell arteritis are fever, inflammatory back pain, diffuse bilateral thigh pain, persistently elevated inflammatory markers, and “atypical polymyalgia rheumatica” (often defined as proximal myalgias with incomplete response to 10 to 15 mg of prednisone daily).37 These patients may be less likely to have cranial ischemic events or vision loss.15
Since aortic involvement is more common than not and symptoms may be subtle or absent, all patients with giant cell arteritis should undergo imaging at baseline to look for and determine the extent of large-vessel involvement.38 Although large-vessel vasculitis may be limited to the aorta, most patients with giant cell arteritis (23 [58%] of 40 in 1 series) also have branch vessel disease at diagnosis, most often in the brachiocephalic and subclavian arteries.15
Patients with giant cell arteritis should routinely undergo an assessment of peripheral pulses and bruits at each visit, and if findings are abnormal or changed they should undergo repeat vascular imaging sooner.
Takayasu arteritis
Takayasu arteritis is a rare systemic large-vessel vasculitis, with an estimated annual incidence of approximately 1 per million.13 In contrast to giant cell arteritis, it is a disease of younger people, with typical onset between ages 15 and 40.13,16 Aortitis is present in the majority of patients with Takayasu arteritis.17–19 Any portion of the aorta can be affected; however, thoracic aortitis is more common in patients from North America or Europe, while abdominal aortitis more frequently occurs in those of Asian ethnicity or with childhood onset.39
The histopathology of Takayasu arteritis aortitis is, in many cases, indistinguishable from that of giant cell arteritis, with granulomatous inflammation and multinucleated giant cells in the medial layer. However, significant hypertrophy of the adventitial layer, when seen, is more typical of Takayasu arteritis.17 Ascending aortic inflammation may lead to aneurysmal change and aortic regurgitation in up to 49% of patients with Takayasu arteritis.40
In contrast to those with giant cell arteritis, patients with Takayasu arteritis may also develop stenoses of the aorta (either in the thoracic or abdominal portions), a radiographic finding peculiar to this form of vasculitis.18 Coronary artery involvement, most typically due to ostial stenosis from an extension of the inflammatory process from the proximal aorta, is also a common and frequently underrecognized manifestation in these young patients.41,42
In addition, branch vessel stenoses are common in the carotid, proximal subclavian, mesenteric, and renal arteries and can lead to lightheadedness, headache, stroke, transient ischemic attack, extremity claudication, postprandial abdominal pain, or early-onset hypertension.19,43 A thorough history is necessary, along with a complete cardiac and peripheral vascular examination, to detect bruits, the murmur of aortic regurgitation, asymmetry or absence of peripheral pulses, and discrepant blood pressures. Complete vascular imaging is required in all patients with suspected Takayasu arteritis for diagnosis and to document the extent of disease. Given the young age of most patients, MRA is the recommended imaging modality; however, CTA or PET-CT can also be used if needed.44
IgG4-related disease
In contrast to giant cell and Takayasu arteritis, IgG4-related disease is a multiorgan fibroinflammatory disease characterized by both tumefactive lesions and, potentially, aortitis. IgG4 aortitis affects about 10% of all patients with IgG4-related disease, at an average age of 58 years, and unlike giant cell arteritis, Takayasu arteritis, and clinically isolated aortitis, is more common in men.20 IgG4-related disease may present as either a true aortitis (with radiographic aortic wall thickening or histopathologic inflammation of tissue or both), or a periaortitis (with significant perivascular inflammation but sparing the vessel wall).21
In 1 series, IgG4-related disease accounted for 3 (9%) of 33 cases of histopathologically identified noninfectious thoracic aortitis, and 3 of the 4 lymphoplasmacytic cases.22 About half of patients with IgG4-aortitis have multiorgan involvement; therefore, clinical evaluation of suspected cases should aim to detect other typical sites of inflammation, such as the salivary glands, lungs, pancreas, biliary tree, and lymph nodes.23
Drug-induced aortitis
Aortitis can also arise after exposure to certain medications, most notably granulocyte colony–stimulating factor25,26 and immune checkpoint inhibitors.27,28 Most patients present with constitutional symptoms (high fever, fatigue, chills) with back, chest, or abdominal pain and significant elevation of inflammatory markers, within days (particularly for granulocyte colony-stimulating factor)25,26 or sometimes months (immune checkpoint inhibitors)27,28 of receiving the drug. Imaging studies are consistent with aortitis in the thoracic or abdominal aorta or both. Recognition is key, as stopping the drug is essential. In many cases, a course of prednisone is also required.25–29
IF ALL OTHER CAUSES ARE RULED OUT: ISOLATED AORTITIS
If infection, primary systemic vasculitides, and other rheumatic diseases are ruled out, and if the inflammation appears limited to the aorta, a diagnosis of isolated aortitis may be made.45 This is a form of single-organ vasculitis in which expression is limited to the aorta, with no features of an underlying systemic vasculitis or rheumatic disease.45 Importantly, isolated aortitis is a diagnosis of exclusion and should be considered a working diagnosis, as its natural history is incompletely understood.
It is important to distinguish whether isolated aortitis was detected histopathologically vs radiographically, as the natural history and approach to treatment may differ. Currently, no universally agreed upon nomenclature exists to distinguish these entities. Pathologists use the term clinically isolated aortitis to refer to aortitis identified incidentally on tissue histopathology after thoracic aortic surgery, in the absence of any identified systemic condition.6 We follow this convention below, while the term isolated aortitis is used to describe aortitis detected radiographically.
Histopathologically detected isolated aortitis (clinically isolated aortitis)
In various retrospective cohort series,4,5,7,8,45,46 noninfectious aortitis was identified histopathologically in 2.5% to 14.6% of thoracic aorta specimens taken during open surgery for aneurysm repair. Interestingly, in most of these series, clinically isolated aortitis was the most common clinical diagnosis, accounting for half to two-thirds of all cases of aortitis.4,8,47,48
Patients with clinically isolated aortitis can be any age, but are typically older (usually in their 60s or 70s),4,5,7 more are female,4,5,7,8 and fewer have concomitant coronary artery disease than those undergoing thoracic aortic surgery for noninflammatory disease (18% vs 45%, P < .01).46 Although most patients are constitutionally well, many (up to 50%) report nonspecific cardiovascular symptoms such as palpitations or dyspnea.4 Because clinically isolated aortitis is typically recognized only postoperatively, many patients do not have blood samples sent to measure their preoperative erythrocyte sedimentation rates or C-reactive protein levels, but when these are available they are usually normal.4,7
Although no single histopathologic pattern defines clinically isolated aortitis, in most cases there is a granulomatous pattern indistinguishable from giant cell arteritis, with a smaller number (5% to 31%) revealing a mixed or lymphoplasmacytic infiltrate.4,5,7
Outcomes in patients with clinically isolated aortitis
The natural history of clinically isolated aortitis (and isolated aortitis) is hard to determine because of the retrospective nature of available data.
Comparing the outcomes of patients with clinically isolated aortitis vs those with other forms of aortitis (giant cell arteritis, Takayasu arteritis, other systemic rheumatic diseases), the risk of subsequent vascular events appears similar across groups. Although patients with clinically isolated aortitis are less likely to develop overt symptoms of vasculitis, new vascular lesions are detected radiographically in about 30% to 45% over time, and 20% to 40% require additional vascular procedures during follow-up.4,5,49 In a series of 217 patients who underwent surgical repair of noninfectious thoracic aortitis, 5 years later 46.7% had either had a vascular complication or died, and 21.8% had undergone a second vascular procedure.5
Interestingly, neither the clinical diagnosis (clinically isolated aortitis vs giant cell arteritis) nor preoperative C-reactive protein level appeared to influence these outcomes, but the segment of the aorta that is involved may matter—arch aortitis was independently associated with an increased risk of vascular complications (hazard ratio [HR] 2.08, P = .005), while descending thoracic aortitis was independently associated with need for a second vascular procedure (HR 2.35), as was aortic dissection (HR 3.08, both P values < .03).5
Over time, 16% to 26% of patients with an initial diagnosis of clinically isolated aortitis may develop overt features of a systemic vasculitis or rheumatic disease.4,5,45 In these cases, the most common new clinical diagnosis is giant cell arteritis/polymyalgia rheumatica, with fewer patients developing features of Takayasu arteritis, spondylarthritis, or IgG4-related disease.
Radiographically detected isolated aortitis (isolated aortitis)
Isolated aortitis is a less common clinical diagnosis in radiographic aortitis series, accounting for only 15% to 23% of all cases (giant cell arteritis is the leading diagnosis).35,36 Although the average age in patients with isolated aortitis is similar to that in patients with clinically isolated aortitis (usually in their 60s),35,36,50 the male-to-female ratio may be increased or even inverted compared with surgical series—9 (82%) of 11 patients were male in 1 series.50
Symptoms of a well-defined systemic rheumatic or vasculitic disease by definition exclude the diagnosis, but in some series up to 50% of patients with isolated aortitis had constitutional symptoms such as fever or weight loss.50 Rather than affecting a focal segment of the thoracic aorta, in most patients the radiographic inflammation extends to multiple aortic sections, with 45% to 82% having abdominal aortitis as well.36,50 In contrast to most surgical clinically isolated aortitis series, erythrocyte sedimentation rates and C-reactive protein values are usually significantly elevated among patients with radiographic isolated aortitis, with typical baseline values between 50 and 100 mg/L and 50 and 100 mm/hour, respectively.35,36
Together, these findings suggest that although patients found to have isolated aortitis based on imaging do not have extension of vasculitis outside of the aorta or clear features of a primary systemic vasculitis such as giant cell arteritis or Takayasu arteritis, they are more likely to have a clinical inflammatory syndrome (constitutional syndrome, elevated inflammatory markers) than those who are diagnosed with clinically isolated aortitis incidentally after surgery, and probably reflect a sicker population.
Outcomes in patients with isolated aortitis
In radiographic series, patients with an initial diagnosis of isolated aortitis appear more likely to have aneurysms or dissections at presentation than those with giant cell arteritis, Takayasu arteritis, or other rheumatic diseases.35 Similarly, a study found that when followed over time, patients with isolated aortitis were significantly more likely to develop new aortic aneurysms than were patients with giant cell arteritis (6 of 44 patients vs 4 of 73 patients, P = .009) and more likely to require aortic surgery (16 vs 10 patients, P = .02) during a median of 34 months of follow-up.36 In another study of noninfectious aortitis in which most cases (77%) were diagnosed radiographically, isolated aortitis was an independent risk factor for subsequent vascular events or vascular procedures.49
Together, these studies suggest that patients with radiographically detected isolated aortitis may experience more severe aortic and vascular outcomes than those with underlying systemic vasculitis or rheumatic disease. It is unclear whether this difference is because patients with isolated aortitis have more aggressive large-vessel inflammation, or because they may come to clinical attention later or are more challenging to recognize than those with cranial or branch vessel manifestations typical of giant cell arteritis or Takayasu arteritis.
I HAVE A PATIENT WITH AORTITIS: WHAT NEXT?
See Figure 3 for an approach to aortitis identified on either histopathology or imaging.4–8,10–29

History and physical examination
One should specifically look for signs and symptoms suggesting an underlying systemic process:6,10–12
Infection (fever, rigors, patient acutely unwell, history of antecedent infection; Table 1)
Large-vessel vasculitis (cranial, ocular, or other vascular ischemia; limb claudication; polymyalgia rheumatica; Table 2)
Variable-vessel vasculitis (most typically oral, genital, ocular, or cutaneous inflammation; Table 2)
Small-vessel vasculitis (ear, nose, throat, pulmonary, renal, cutaneous, or nerve involvement; Table 2)
Other rheumatic diseases that may have concomitant aortic involvement, such as rheumatoid arthritis, spondyloarthritides, sarcoidosis, IgG4-related disease, Cogan syndrome, relapsing polychondritis, systemic lupus erythematosus, or other systemic autoimmune diseases (Table 3).
A thorough medication history is also critical to exclude drug-induced aortitis, which has been reported with granulocyte-colony stimulating factor25,26 and immune checkpoint inhibitor exposure.27–29
Laboratory tests
At minimum, all patients with noninfectious aortitis should have the following laboratory tests:10–12
Complete blood cell count with differential
Serum creatinine level and urinalysis (and protein quantification if proteinuria is detected)
Calcium level
Albumin level
C-reactive protein level
Erythrocyte sedimentation rate
Blood cultures.
Testing to exclude tuberculosis or syphilis exposure is also appropriate for most. Antinuclear antibody, anti-neutrophil cytoplasmic antibodies, extractable nuclear antigen, anti-double-stranded DNA, complement components 3 and 4, serum IgG4 level, or human leukocyte antigen typing (B27 or B51) may be considered.
After infection is excluded, all patients with thoracic aortitis should be referred to a vasculitis expert for assessment.
Imaging studies
Complete imaging of the entire aorta and branch vessels (skull base through thighs) at diagnosis is essential in all patients to document the extent of vascular disease already present and to serve as a baseline for comparison over time.4 CTA, MRA, or PET-CT can be used, depending on patient factors and access.30
In certain conditions, PET-CT may show additional asymptomatic sites of FDG avidity outside of the aorta that may strongly suggest a particular diagnosis (eg, sinus, lung, and hilar lymph node avidity in sarcoidosis vs submandibular, lacrimal gland, and pancreatic avidity in IgG4-related disease).21 These additional sites of activity may be safer sites to obtain confirmatory biopsy specimens in cases in which aortitis is diagnosed radiographically.
APPROACH TO TREATMENT
After aortitis is diagnosed, whether to initiate immunosuppression depends largely on the patient’s clinical picture, how the aortitis was detected, and whether there is evidence or suspicion of persistent active vasculitis. For patients with a systemic vasculitic disease such as giant cell arteritis or Takayasu arteritis, immunosuppression with prednisone (with or without other agents; see “Choice of steroid-sparing therapy” below) is almost always warranted.
The benefits vs risks of systemic immunosuppression for patients with disease limited to the aorta, however, must be considered.
Treatment of clinically isolated aortitis
In surgical series, only a minority of patients with clinically isolated aortitis were treated with glucocorticoids postoperatively, and results were mixed.4,46,51
In 1 study, new vascular lesions were noted to develop in fewer patients with clinically isolated aortitis who received prednisone postoperatively (2 of 11, 18%) than in those who did not (27 of 54, 50%), raising a possibility of benefit.4 In another study, of 23 patients with clinically isolated aortitis (of whom 11 received postoperative corticosteroids), the potential of harm was raised when those receiving treatment were found to have a nonsignificantly increased growth rate of aneurysmal dilation in the descending aorta during follow-up.51
In the study of 217 patients with noninfectious thoracic aortitis (most with clinically isolated aortitis phenotype), no association was observed between use of postoperative glucocorticoids or other immunosuppressive treatment and vascular outcomes. Intriguingly, however, use of statins was independently associated with reduced likelihood of a subsequent vascular procedure (HR 0.47, 95% confidence interval 0.24–0.90, P = .028).5 This finding deserves further study.
Ultimately, whether to initiate glucocorticoids in patients with clinically isolated aortitis identified histopathologically after surgery is controversial, and the decision often balances on whether symptoms or signs of active vascular inflammation persist, and should be made on an individual basis.
Treatment of isolated aortitis
In radiographically identified isolated aortitis, because the aortitis remains in situ, once vascular inflammation is identified, most patients require immunosuppression, regardless of clinical phenotype.35,36,49 Due to its rapid onset of action, prednisone is typically used first-line. In some series, prednisone monotherapy was favored.36,49 However, in view of the high risks of subsequent vascular events and the known toxicities of glucocorticoids, some authors describe early use of steroid-sparing therapy.35
Choice of steroid-sparing agents
Methotrexate is the most frequently used steroid-sparing agent,35 but other options include azathioprine, mycophenolate mofetil, tocilizumab, rituximab, and leflunomide. Decisions on whether to add a steroid-sparing agent and which agent to use are influenced by the underlying cause of aortitis, as well as comorbidities and patient preference. Although there is no evidence to guide this decision in clinically isolated aortitis and isolated aortitis, in giant cell arteritis best evidence supports the early use of tocilizumab in combination with glucocorticoids, or methotrexate if tocilizumab is not available or is contraindicated.38 Methotrexate, azathioprine, and antitumor necrosis factor alpha therapy used early in combination are excellent choices for Takayasu arteritis, while rituximab is preferred for IgG4-related aortitis when a second agent is required.52
Management of traditional cardiovascular risk factors
Traditional cardiovascular risk factors, including hypertension, dyslipidemia, and smoking, are common, present in one-quarter to two-thirds of patients with aortitis.5 Due to the high risk of subsequent vascular events, blood pressure, lipid profile, blood glucose, and smoking status should be assessed in all patients. Counseling regarding lifestyle modification should be offered, and biochemical risk factors aggressively treated.39,53
Monitoring recommendations
Large-vessel vasculitis can progress over time, even without clinical findings or elevated inflammatory markers, both in patients with isolated aortitis or clinically isolated aortitis and those with recognized systemic large-vessel vasculitides.4,43 Given the high frequency of new vascular lesions and need for repeat vascular procedures over time, it is advised that all patients with aortitis be monitored with serial imaging of the entire aorta and major branch vessels.4,10,11 There is no consensus on the frequency with which imaging be repeated, but 1 center recommends it be done yearly (or sooner if clinical parameters suggest progression).4
SUMMARY AND RECOMMENDATIONS
Aortitis is a manifestation of a heterogenous group of diseases, so one approach does not fit all. Once it is detected, either histopathologically or radiographically, patients with aortitis require a full clinical assessment, basic laboratory tests, and complete vascular imaging of the entire aorta and major branches. These patients should be referred to a vasculitis expert to help guide the workup and determine the clinical diagnosis.
The decision to initiate immunosuppression is guided by the clinical assessment, including the presence or absence of an active systemic condition, whether active vascular inflammation persists, and the risks and benefits to the individual patient. Given the high risk of developing subsequent vascular lesions or requiring additional vascular procedures, regardless of treatment, all patients with aortitis should have aggressive management of traditional cardiovascular risk factors and be followed with serial clinical assessments, inflammatory markers, and large-vessel imaging. Team-based care may help guide treatment decisions in these complex cases.
DISCLOSURES
Dr. Clifford has disclosed serving as a research site principal investigator for Abbvie Pharmaceuticals, teaching and speaking for UCB, and serving as a research site subinvestigator for UCB.
- Copyright © 2024 The Cleveland Clinic Foundation. All Rights Reserved.
![]()
Clicking the link below will connect you to begin the credit-claiming process for CME and MOC. After clicking on the link, scroll to the bottom of the page and click on “Complete the CME/MOC Process.” You will need your myCME login information to access this.




{kind=link}
{kind=link}
{kind=link}