


ABSTRACT
Monoclonal gammopathy of undetermined significance (MGUS) is commonly diagnosed in outpatients being worked up for an array of clinical concerns. It carries a risk of progression to myeloma and other lymphoproliferative disorders that, albeit low (1% per year), warrants regular follow-up. Patients with MGUS can be risk-stratified on the basis of the amount and type of their monoclonal protein as well as whether they have an abnormal light-chain ratio. Here, we provide a guide to the diagnosis, workup, and management of MGUS.
MGUS is the most common of the monoclonal gammopathies.
The overall risk of MGUS progressing to myeloma and other lymphoproliferative disorders is 1% per year.
Low-risk MGUS is defined by an immunoglobulin G monoclonal protein at a concentration less than 1.5 g/dL and a normal serum free light-chain ratio.
Low-risk MGUS carries a much lower risk of progression than intermediate- and high-risk MGUS, may not require subspecialty referral, and can be followed by the outpatient provider.
The monoclonal gammopathies encompass a number of disorders characterized by the production of a monoclonal protein (M protein) by an abnormal clone of plasma cells or other lymphoid cells. Monoclonal gammopathy of undetermined significance (MGUS) is the most common of these disorders. The diagnostic criteria for MGUS are listed in Table 1.
Diagnostic criteria for MGUS, smoldering multiple myeloma, and active multiple myeloma
Its clinical relevance lies in the inherent risk of progression to hematologic malignancies such as multiple myeloma or other lymphoproliferative disorders, or of organ dysfunction due to the toxic effects of the M protein. An M protein may consist of an intact immunoglobubin (Ig) molecule–ie, 2 light chains and 2 heavy chains (most commonly IgG type followed by IgA and IgM)–or a light chain only (kappa or lambda) (Figure 1).
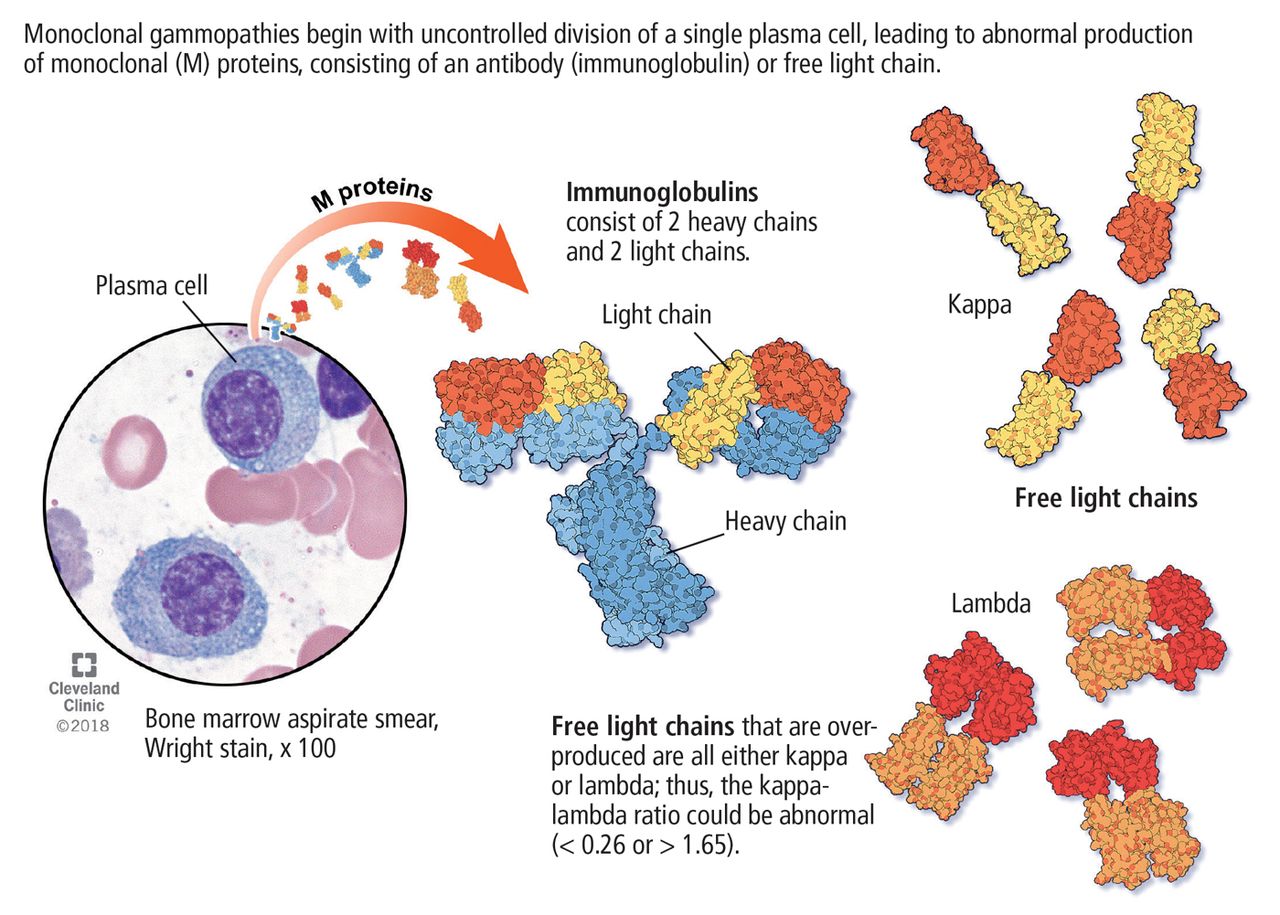
Monoclonal gammopathies begin with uncontrolled division of a single plasma cell, leading to abnormal production of monoclonal (M) proteins, consisting of an antibody (immunoglobulin) or free light chain.
MGUS is present in 3% to 4% of the population over age 50 and is more common in older men, African Americans, and Africans.1–6
The overall risk of progression to myeloma and related disorders is less than or equal to 1% per year depending on the subtype of the M protein (higher risk with IgM than non-IgM and light-chain MGUS).7,8 While the risk of malignant transformation is low, multiple myeloma is almost always preceded by the presence of an asymptomatic and often unrecognized monoclonal protein.
WHEN SHOULD WE LOOK FOR AN M PROTEIN?
An M protein is typically an incidental finding when a patient is being assessed for any of a number of presenting symptoms or conditions. A large retrospective study9 found that screening for MGUS was mostly performed by internal medicine physicians. The indications for testing were anemia, bone-related issues, elevated creatinine, elevated erythrocyte sedimentation rate, and neuropathy.
Routine screening for an M protein in the absence of clinical suspicion is not recommended, given the low risk of malignant progression, lack of effect on patient outcomes, the accompanying emotional burden, and lack of treatment options.5,10 Evaluation for monoclonal gammopathy may be considered as part of the workup of associated clinical symptoms and signs and laboratory and imaging findings (Table 2).2,10,11
Indications for testing for monoclonal gammopathy
A low anion gap is not a major indicator of an M protein unless in a high concentration, in which case other manifestations would be present, such as renal failure, which would guide the diagnosis. Polyclonal hypergammaglobulinemia as a cause of low anion gap is far more common than MGUS.
HOW SHOULD WE SCREEN FOR AN M PROTEIN?
Serum protein electrophoresis is an initial test used to identify an M protein and has a key role in quantifying it (Figure 2). An M protein appears as a narrow spike on the agarose gel and should be distinguished from the broad band seen in polyclonal gammopathies associated with cirrhosis and chronic infectious and inflammatory conditions, among others.12 A major disadvantage of serum protein electrophoresis is that it cannot detect an M protein in very low concentrations or determine its identity.

Serum protein electrophoresis from a patient with monoclonal gammopathy of undetermined significance (right) shows an abnormal band of gamma globulin (labeled M) that is not present in a normal study (left).
Serum immunofixation is more sensitive than serum protein electrophoresis and should always be ordered in conjunction with it, mostly to ensure detecting tiny amounts of M protein and to identify the type of its heavy chain and light-chain components.13
The serum free light-chain assay is also considered an essential part of the screening process to detect light-chain MGUS and light-chain myeloma. As many as 16% of myeloma patients secrete only light chains, which may not be identified on serum immunofixation.3,6,7,10,14,15 In general, a low kappa-lambda ratio (< 0.26) indicates the overproduction of lambda light chains, and a high ratio (> 1.65) indicates the overproduction of kappa light chains.
The serum free light-chain assay helps detect abnormal secretion of monoclonal light chains before they appear in the urine once the kidney tubules become saturated and unable to reabsorb them.
Of note, the free light-chain ratio can be abnormal (< 0.26 or > 1.65) in chronic kidney disease. Thus, it may be challenging to discern whether an abnormal light-chain ratio is related to impaired light-chain clearance by the kidneys or to MGUS. In general, kappa light chains are more elevated than lambda light chains in chronic kidney disease, but the ratio should not be considerably skewed. A kappa-lambda ratio below 0.37 or above 3 is rarely seen in chronic kidney disease and should prompt workup for MGUS.16
Tests in combination. The sensitivity of screening for M proteins ranges from 82% with serum protein electrophoresis alone to 93% with the addition of serum immunofixation and to 98% with the serum free light-chain assay.15 The latter can replace urine protein electrophoresis and immunofixation when screening for M protein, given its higher sensitivity.15,17 An important caveat is that urine dipstick testing does not detect urine light chains.
Once an M protein is found, immunoglobulin quantification, a complete blood cell count, and serum creatinine and calcium measurements are also recommended to look for anemia, renal failure, and hypercalcemia, which can be associated with symptomatic myeloma.3,5,6,18–22
Table 3 lists the initial laboratory tests required in patients with MGUS.
Initial laboratory tests in MGUS
WHAT IS THE DIFFERENTIAL DIAGNOSIS OF MONOCLONAL GAMMOPATHIES?
MGUS should be differentiated from other plasma-cell and lymphoproliferative disorders that feature an M protein and would otherwise require treatment (Table 4). The differential diagnosis includes smoldering multiple myeloma, symptomatic multiple myeloma, Waldenström macroglobulinemia, lightchain amyloidosis, low-grade B-cell lymphoproliferative disorders, a variety of monoclonal protein-related kidney disorders, and plasmacytomas.10,14
Monoclonal gammopathy: Differential diagnosis
MGUS
Based on the International Myeloma Working Group consensus, a formal diagnosis of MGUS is established when a serum M protein is detected and measured at a concentration less than 3 g/dL on serum protein electrophoresis along with less than 10% clonal plasma cells in the bone marrow.1–6,14,18,19 Nevertheless, bone marrow biopsy can be omitted in certain patients as discussed below. The absence of myeloma-related organ damage– particularly osteolytic bone lesions, anemia, otherwise unexplained renal failure, and hypercalcemia–is fundamental and necessary for a diagnosis of MGUS.
Smoldering multiple myeloma
Compared with patients with MGUS, patients with smoldering multiple myeloma have higher M protein concentrations (≥ 3 g/dL) or 10% or more clonal plasma cells in the marrow or both, and are at higher risk of progression to symptomatic multiple myeloma. Nevertheless, like patients with MGUS, they have no myeloma symptoms or evidence of end-organ damage.
Symptomatic multiple myeloma
By definition, patients with multiple myeloma develop organ damage related to their malignancy and need therapy to halt disease progression. Multiple myeloma causes clinical manifestations through cellular infiltration of the bone and bone marrow (anemia, osteolysis, and hypercalcemia) and light chain-induced toxicity (renal tubular damage and cast nephropathy).
In 2014, the definition of multiple myeloma was updated to include 3 new myeloma-defining events that herald a significantly higher risk of progression from smoldering to symptomatic multiple myeloma, and now constitute an integral part of the diagnosis of symptomatic multiple myeloma. These are:
Focal lesions (> 1 lesion larger than 5 mm) visible on magnetic resonance imaging
≥ 60% clonal plasma cells on bone marrow biopsy
Ratio of involved to uninvolved serum free light chains ≥ 100 (the involved light chain is the one detected on serum protein electrophoresis and immunofixation).14
Bone pain, symptoms of anemia, and decreased urine output may suggest myeloma, but are not diagnostic. Although the “CRAB” criteria (elevated calcium, renal failure, anemia, and bone lesions) define multiple myeloma, the presence of anemia, hypercalcemia, or renal dysfunction do not by themselves mark transformation from MGUS to multiple myeloma. Thus, other causes need to be considered, since the risk of transformation is so low. Importantly, hyperparathyroidism must be ruled out if hypercalcemia is present in a patient with MGUS.10
Waldenström macroglobulinemia
Waldenström macroglobulinemia, also called lymphoplasmacytic lymphoma, is an indolent non-Hodgkin B-cell lymphoma that can invade the marrow, liver, spleen, and lymph nodes, leading to anemia and organomegaly. It features a monoclonal IgM protein that can be associated with increased blood viscosity, cold agglutinin disease, peripheral neuropathy, and cryoglobulinemia.
Waldenström macroglobulinemia should be suspected in any patient with IgM type M protein and symptoms related to hyperviscosity (headache, blurry vision, lightheadedness, shortness of breath, unexplained epistaxis, gum bleeding); systemic symptoms (fever, weight loss, and night sweats); and abdominal pain (due to organomegaly).23
Monoclonal gammopathy of renal significance
Monoclonal gammopathy of renal significance (MGRS) is a newly recognized entity defined by kidney dysfunction associated with an M protein without evidence of myeloma or other lymphoid disorders.24 Multiple disorders have been included in this category with different underlying mechanisms of kidney injury. This entity is beyond the scope of this discussion.
Light-chain amyloidosis
Misfolded light-chain deposition leading to organ dysfunction is the hallmark of light-chain amyloidosis, which constitutes a subset of MGRS. An abnormal light-chain ratio, especially if skewed toward lambda should trigger an investigation for light-chain amyloidosis.10
Abnormal light chains may infiltrate any organ or tissue, but of greatest concern is infiltration of the myocardium with ensuing heart failure manifestations. N-terminal pro-B-type natriuretic peptide (NT-proBNP) is a sensitive marker for cardiac amyloidosis in the presence of suggestive features on transthoracic echocardiography (eg, left ventricular hypertrophy) but is not specific as it can be elevated in heart failure regardless of the underlying cause.10
Glomerular injury with nephrotic syndrome may also point toward renal involvement by light-chain amyloidosis and establishes a key distinctive factor from myeloma in which tubular injury is the main mechanism of kidney dysfunction.
Clinical clues for light-chain amyloidosis include heart failure symptoms, neuropathy, and macroglossia. If any of these symptoms and signs is present, we recommend electrocardiography (look for low voltage in limb leads), transthoracic echocardiography, measuring the NT-proBNP level, and urinalysis to look for albuminuria. Notably, carpal tunnel syndrome may be a very early clinical manifestation of amyloidosis, but by itself it is nonspecific. Light-chain amyloidosis is a common cause of macroglossia in adults.10,25
Neuropathy associated with M proteins is a clinical entity related to a multitude of disorders that may necessitate treating the underlying cellular clone responsible for the secretion of the toxic M protein. These disorders include light-chain amyloidosis, POEMS (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes or sclerotic bone lesions) syndrome, and IgM-related neuropathies with anti-myelin-associated glycoprotein antibodies.3,10,11,14
Notably, weight loss and fatigue in a patient with MGUS may be the first signs of light-chain amyloidosis or Waldenström macroglobulinemia and should prompt further evaluation.25
HOW ARE PATIENTS WITH MGUS RISK-STRATIFIED AND FOLLOWED?
Research has helped to refine the diagnostic workup and recognize subsets of patients with MGUS at different risks of progression to myeloma and related disorders. Factors predicting progression are1,6,7,26,27:
The amount of the M protein
The type of M protein (IgG vs non-IgG)
An abnormal free light-chain ratio. Based on these predictors, MGUS can be classified into 4 risk categories: low, low-intermediate, high-intermediate, and high (Table 5).
Risk factors for progression in MGUS
Half of patients with MGUS fall into the low-risk category, which is defined by IgG-type serum M protein in a concentration less than 1.5 g/dL and a normal serum free light-chain ratio (kappa-lambda 0.26–1.65).5,27 The absolute risk of progression at 20 years is only 5% for patients with low-risk MGUS, compared with 58% in patients with high-risk MGUS (positive for all 3 risk factors).5
The presence of less than 10% plasma cells in the bone marrow is required to satisfy the definition of MGUS, but bone marrow biopsy can be omitted for patients with low-risk MGUS, given the slim chance of finding a significant percentage of clonal plasma cells in the marrow and the inherently low risk of progression.5,10 Skeletal surveys are often deferred for low-risk MGUS, but we obtain them in all our patients to ensure the absence of plasmacytomas, which need to be treated (typically with radiotherapy). Importantly, patients with unexplained bone pain (mostly in long bones, ribs, and spine, whereas joints are not typically involved) and a normal skeletal survey should undergo advanced imaging (whole-body magnetic resonance imaging or whole-body positron emission tomography and computed tomography) to detect bone lesions otherwise missed on plain radiography.28,29
Most of the recommendations regarding follow-up are based on expert opinion, given the lack of randomized data. Most experts agree that all patients should be reevaluated 6 months after an M protein is detected, with laboratory surveillance tests (complete blood cell count, serum creatinine, serum calcium level, serum protein electrophoresis, and serum free light chains). Low-risk patients with a stable M protein level can be followed every 2 to 3 years.
Suspect malignant progression if the serum M protein level increases by 50% or more (with an absolute increase of ≥ 0.5 g/dL); the serum M protein level is 3 g/dL or higher; the serum free light-chain ratio is more than 100; or the patient has unexplained anemia, elevated creatinine, bone pain, fracture, or hypercalcemia.
Patients at intermediate or high risk should be followed annually after the initial 6-month visit.5,7,10
A recent study highlighted the importance of risk stratification in reducing the costs associated with an overzealous diagnostic workup of patients with low-risk MGUS.30 These savings are in addition to a reduction in patient anticipation and anxiety that universally occur before invasive procedures.
THE ROLE OF THE PRIMARY CARE PROVIDER AND THE HEMATOLOGIST
Once an M protein is identified, a comprehensive history, physical examination, and laboratory tests (serum protein electrophoresis to quantify the protein, serum immunofixation, serum free light chains, complete blood cell count, calcium, and creatinine) should be done, taking into consideration the differential diagnosis of monoclonal gammopathies discussed above. After MGUS is confirmed, the patient should be risk-stratified to determine the need for bone marrow biopsy and to predict the risk of progression to more serious conditions.
Referral to a hematologist is warranted for patients with intermediate- and high-risk MGUS, patients with abnormal serum free light-chain ratios, and those who show evidence of malignant progression. Patients with intermediate-and high-risk MGUS could be referred for bone marrow biopsy before assessment by a hematologist. The primary care provider may continue to follow patients with low-risk MGUS who do not display clinical or laboratory evidence of myeloma or related disorders.
When light-chain amyloidosis, Waldenström macroglobulinemia, or another M protein-related disorder is suspected, referral to subspecialists is advised to better define the correlation between the M protein and the pa tient’s symptoms and signs (Table 6).
MGUS: When to refer patients to a hematologist
The importance of educating patients to report any new worrisome symptom (eg, fatigue, neuropathy, weight loss, night sweats, bone pain) cannot be over-emphasized, as some patients may progress to myeloma or other disorders between follow-up visits.
Footnotes
Dr. Valent has disclosed teaching and speaking for Amgen, Celgene, and Takeda.
- Copyright © 2019 The Cleveland Clinic Foundation. All Rights Reserved.




{kind=link}
{kind=link}