


A 52-year-old woman presented to the emergency department in Las Vegas, NV, with progressively worsening altered mental status for the past 2 weeks. A history of symptoms was obtained from the patient’s sister, with whom she was staying. The sister reported that during the week before presentation, the patient had episodes of confusion, intermittent blank staring, blurred vision, bilateral incoordination of the upper and lower extremities, difficulty following commands, and decreased verbal communication. The patient had also been holding her left hand in a fist. Two days before presentation, the patient experienced a significant decline in mentation and had multiple episodes of urinary incontinence, which she never had before. During a period of lucidity in the emergency department, the patient denied having fever, chills, nausea, vomiting, chest pain, shortness of breath, abdominal pain, dysuria, or headache.
The patient had a history of major depressive disorder treated with fluoxetine until 8 days before presentation, when she was switched to escitalopram by an outpatient psychiatrist owing to onset of the psychomotor symptoms. The patient had no prior blood transfusions or surgeries and no known drug allergies.
The patient was from California and spoke only Spanish. She was employed by a shoe store, was single, lived alone, and had no children. She had been fully independent in activities of daily living, maintained full-time employment, and was financially stable, but did not have health insurance. She did not smoke, drink alcohol in excess, or use recreational drugs.
On physical examination, the patient was in moderate distress and exhibited waxing and waning alertness. She was consistently arousable with painful stimulation. She was aware that she was in a hospital that was not in California but was otherwise disoriented. Her Glasgow Coma Scale score was 14 out of 15, ie, responsive (range 3–15, with 3 being completely unresponsive). Her blood pressure was 148/76 mm Hg, heart rate 89 beats per minute, body temperature 36.7°C (98.0°F), and respiratory rate 20 breaths per minute, and the oxygen saturation level was 97% on room air. Body mass index was 30 kg/m2.
Cardiovascular, pulmonary, and abdominal examinations were normal. The head was normocephalic and atraumatic, with anicteric sclera and moist mucous membranes. Pupils were equal, round, and reactive to light, and extraocular muscles were grossly intact.
On neuropsychiatric examination, the patient had poor concentration and difficulty participating. She had frequent episodes of staring into space with periods of rhythmic jerking of the eyes, head, and bilateral upper extremities (opsoclonus and myoclonus). During lucid intervals, she demonstrated intact cranial nerves II to XII and did not show facial asymmetry, gaze preference, inappropriate saccades, nystagmus, or dysarthria. A Babinski reflex test revealed downgoing toes bilaterally. However, significant spasticity and resistance to range of movements were noted, along with 5 to 7 beats of ankle clonus bilaterally after passive dorsiflexion.
Cerebellar and gait examinations were deferred because of the patient’s inability to follow commands. Also, we could not perform a Mini-Mental State Examination or Montreal Cognitive Assessment. The patient was admitted for further evaluation.
1. Which of the following would be an atypical cause of this patient’s rapidly progressive cognitive decline?
Stroke
Toxic metabolic encephalopathy
Infectious encephalomyelitis
Psychosis
Alzheimer disease
Malignancy
DIFFERENTIAL DIAGNOSIS: DISEASES, CONDITIONS TO RULE OUT
The initial differential diagnosis for rapidly progressive cognitive decline includes vasculopathy, toxic metabolic encephalopathy, infectious encephalomyelitis, malignancy, and neurodegenerative and psychiatric causes.
Although this patient’s myoclonus, progressive encephalopathy, and waxing and waning alertness would be an atypical presentation for stroke, this should be ruled out first because of the potential for rapid, irreversible ischemia to neural tissue. If diagnosed correctly and early enough, acute stroke can be treated either with antiplatelet therapy for ischemic stroke or with surgical or endovascular management for hemorrhagic stroke.
Drug-induced encephalopathies due to lithium, amitriptyline, and baclofen, heavy metal intoxication (eg, bismuth subsalicylate, manganese), and metabolic encephalopathies such as Wernicke-Korsakoff syndrome and vitamin B12 deficiency are all possible causes of rapidly progressive dementia. Given the patient’s abrupt transition off fluoxetine before admission, serotonin syndrome is also a consideration.
There are several infectious causes of rapid cognitive decline. These include Whipple disease (subacute dementia, ataxia, and myoclonus), human immunodeficiency virus (HIV) encephalitis, tuberculosis, herpes simplex encephalitis, and subacute sclerosing panencephalitis.1,2 While not strictly caused by infectious pathogens, spongiform encephalopathies (also called prion diseases) such as fatal familial insomnia, kuru, and Creutzfeldt-Jakob disease (CJD) can present with rapid cognitive decline. Autoimmune etiologies should also be considered, including antibodies to both extracellular antigens such as N-methyl-D-aspartate receptor and intracellular antigens such as Hu antigens.
Common causes of chronic cognitive decline include neurodegenerative diseases such as Alzheimer disease and frontotemporal dementia (Pick disease), and dementia associated with movement disorders, such as Parkinson disease, Lewy body dementia, and Huntington disease. Although cognitive decline typically occurs over years in these diseases, atypical presentations can lead to rapidly progressive dementia in 15% to 25% of cases and thus should be considered in such cases.3
Vascular dementia (eg, multi-infarct dementia, subcortical arteriosclerotic encephalopathy) should be considered in patients with a history of or risk factors for atherosclerotic vascular disease such as diabetes mellitus and hypertension, and risk factors for thromboembolism such as atrial fibrillation and endocarditis. Additional vascular causes could be autoimmune or inflammatory in nature, including primary central nervous system vasculitis or Susac syndrome. Processes that result in mass-effect central nervous system changes can facilitate acute or chronic cognitive decline. These include tumor and cyst, and disorders of cerebrospinal fluid (CSF) production or outflow, such as normal pressure hydrocephalus.
Initial diagnostic workup
In patients with rapid cognitive decline, the initial workup includes a variety of imaging and laboratory testing. Imaging should include urgent computed tomography (CT) of the head without contrast to assess for hemorrhage and mass effect, magnetic resonance imaging (MRI) of the brain, vascular imaging of the head and neck (options include magnetic resonance angiography, CT angiography, and ultrasonography), and echocardiography. Initial laboratory tests should include complete blood cell count, serum thyroid-stimulating hormone, electrolytes (including sodium, calcium, blood urea nitrogen, and creatinine), liver enzyme tests, and toxicology screening. If imaging or the history does not indicate stroke, additional testing can include thiamine and vitamin B12 (cobalamin) levels, serologic testing for HIV, hepatitis, and syphilis, and CSF studies, including glucose, protein, gram stain, and culture. These tests can rule out most reversible causes of rapidly progressive dementia, such as infection or metabolic abnormalities.4
The most common first-line test for vitamin B12 deficiency is the serum vitamin B12 level, which has reasonable sensitivity and is widely available and relatively inexpensive. The serum methylmalonic acid level, the standard confirmatory test, has both higher sensitivity and specificity than serum vitamin B12 and can be used to track treatment response, but it is less widely available and more expensive. In practice, it is not unreasonable to order both tests at once if resources allow and if pretest probability for vitamin B12 deficiency is moderate or high.5,6
CASE CONTINUED
Based on the patient’s presentation, several potential diagnoses were ruled out. Serotonin syndrome was determined to be unlikely because her symptoms were more progressive in onset (> 24 hours) and began before switching to escitalopram. This was further supported by lack of spontaneous clonus, diaphoresis, agitation, hyperreflexia, or body temperature above 38°C (100.4°F).
The complete blood cell count results showed no leukocytosis, a mean cellular volume of 89.7 fL (reference range 80.1–98.4), and a hemoglobin of 14.8 g/dL (11.0–14.9). An electrolyte panel was normal except for hypokalemia, with a level of 2.9 mmol/L (3.5–5.0). Phosphate and magnesium levels were normal. Results from tests for thyroid function, liver function, vitamin B12, thiamine, and vitamin E were all within normal limits. A urine toxicology screen was negative. Microbiological screening assay results were negative for HIV-1 and HIV-2, viral hepatitis, syphilis, and Lyme disease.
In the likely absence of other infectious causes, fungal infection was considered. However, a beta-D-glucan assay for invasive or disseminated fungal infections was negative.
Imaging studies
CT of the head without contrast was negative for edema, herniation, hemorrhage, and ventriculomegaly, effectively ruling out mass effect or normal pressure hydrocephalus.
Brain MRI showed cortical bifrontal and parietal diffusion restriction on diffusion-weighted imaging—a finding also called cortical ribboning (Figure 1). No thalamic hyperintensity was noted. No multiple infarcts suggestive of progressive vascular dementia were seen; these are typically seen with primary angiitis of the central nervous system or uncontrolled hypertension or diabetes.
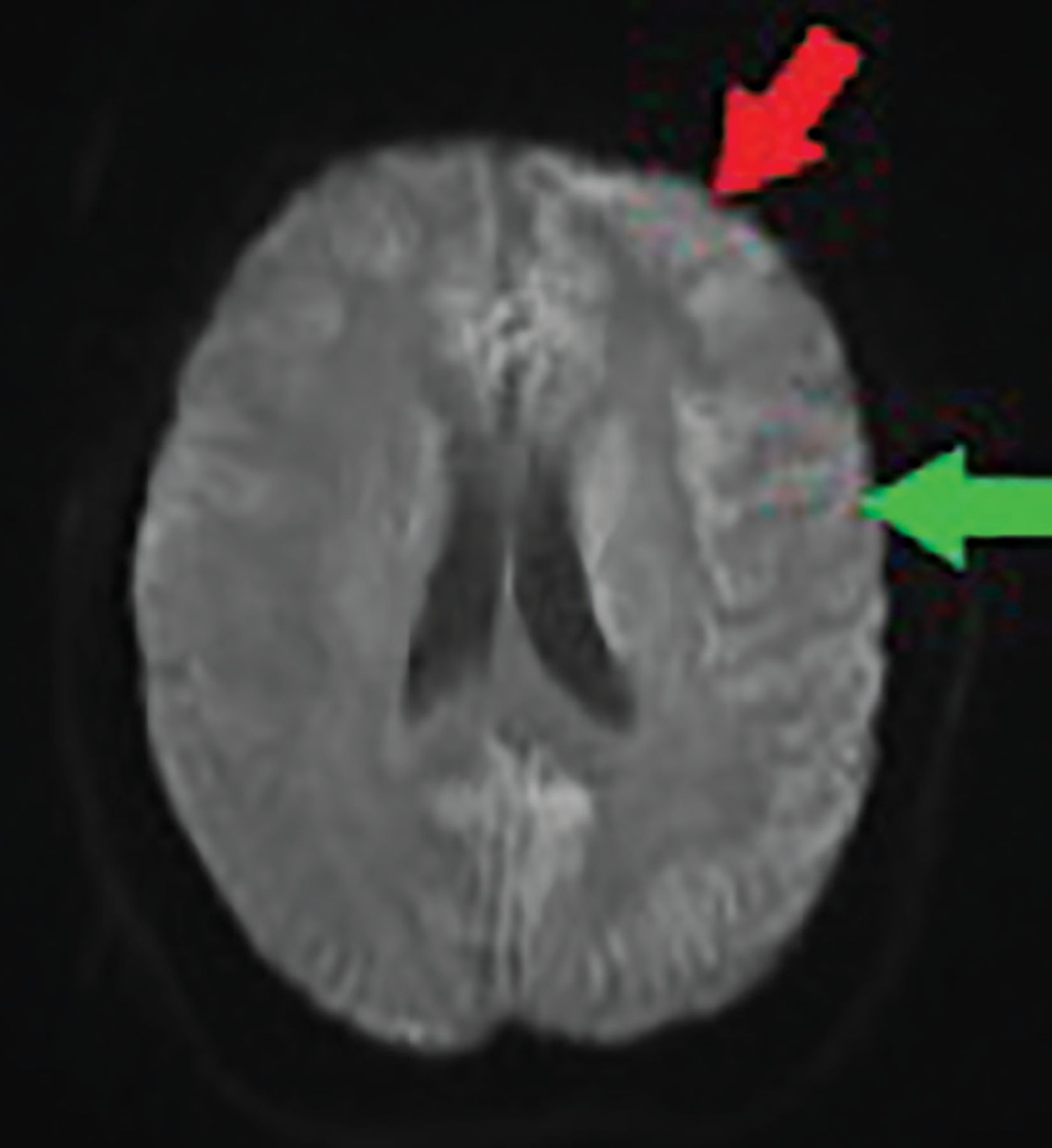
Diffusion-weighted magnetic resonance imaging at admission shows cortical bifrontal (red arrow) and parietal (green arrow) diffusion (cortical ribboning), with greater intensity on the left. No thalamic hyperintensity was seen. Note that the quality of this image was affected by patient movement during the procedure, in spite of attempts to sedate her.
CT angiography of the head and neck to further assess for vascular causes (ie, primary central nervous system vasculitis) noted no vascular abnormality. Echocardiography to investigate a source of possible embolus showed normal left ventricular function and no valvular pathology or thrombus. Electrocardiography showed normal sinus rhythm.
Additional laboratory testing
CSF testing showed clear, colorless fluid, glucose within normal limits, white blood cell count of 3 × 109/L (reference range 5–10), and red blood cell count of 19 × 1012/L (4.2–6.1). No oligoclonal bands or organisms were seen, and CSF cultures were negative. Results from a CSF Venereal Disease Research Laboratory test and 14-pathogen meningitis polymerase chain reaction panel were also negative. (The meningitis panel detects 14 bacterial, viral, and fungal pathogens: Escherichia coli K1, Haemophilus influenzae, Listeria monocytogenes, Streptococcus agalactiae, Streptococcus pneumoniae, cytomegalovirus, enterovirus, herpes simplex virus 1 and 2, human herpesvirus 6, human parechovirus, varicella zoster virus, and Cryptococcus neoformans/gatti.)
Both the CSF protein and myelin basic protein were mildly elevated: CSF protein 65 mg/dL (14–40), myelin basic protein 6.7 ng/mL (0.0–1.2). While these CSF findings can be suggestive of multiple sclerosis,7,8 the clinical presentation and MRI findings did not suggest multiple sclerosis and oligoclonal bands were negative, effectively ruling out this diagnosis. As imaging findings were not consistent with progressive multifocal luekoencephalopathy and there was no history of immunosuppression, the John Cunningham viral polymerase chain reaction test was not performed on the CSF sample. A CSF analysis for diagnostic markers of Alzheimer disease, including CSF total amyloid, hyperphosphorylated tau (p-tau), and tau-tau ratio, also was not conducted during the initial evaluation, since rapidly progressive Alzheimer disease typically takes months to years to evolve, as opposed to our patient’s 2-week decline.9,10
The bottom line
The patient’s initial radiologic and laboratory results were largely unremarkable, except for mild CSF protein elevation and cortical ribboning on brain MRI, significant progressive cognitive decline, and myoclonus. Therefore, our focus shifted to less common causes of altered mental status, including antibody-mediated encephalitis (50–80 cases per million people per year, according to a prospective study in England),11,12 paraneoplastic syndrome, and prion diseases including CJD (1–1.5 cases per million population per year).13
AUTOANTIBODY ENCEPHALITIS: WHAT TO LOOK FOR
Antibody-mediated encephalitis
Antibody-mediated (or autoimmune) encephalitis is a class of disorders caused by antibodies typically directed against cell-surface antigens located on various components of the central nervous system. They are characterized by a wide range of neuropsychiatric symptoms, including behavioral changes, seizures, and abnormal movements. Autoantibody encephalitis can occur at all ages.
Cases typically resolve partially or completely with appropriate diagnosis and treatment. Treatment is focused on removal of any identifiable triggers (eg, tumor) and immunosuppression with systemic glucocorticoids, intravenous immunoglobulin, or plasmapheresis.
While there are more than 15 antibody-associated encephalitides that have been identified, the most common subtypes are anti-N-methyl-D-aspartate receptor antibody encephalitis (incidence of 1.5 cases per million per year) and leucine-rich glioma-inactivated 1 autoantibody encephalitis (0.8 cases per million per year according to a retrospective Dutch study).12,14
Paraneoplastic syndromes
Paraneoplastic syndromes are the result of immune-mediated damage from malignancy-associated antibodies directed against intracellular proteins. Commonly implicated antibodies in paraneoplastic encephalitis include anti-Hu, anti-Yo, and anti-Ma antibodies.
Signs and symptoms depend on the affected component of the nervous system and may include confusion, myoclonus, and ataxia. It is not uncommon for patients to initially seek medical care for paraneoplastic symptoms resulting from an undiagnosed malignancy.15
As with the antibody-mediated encephalitides, treatment is focused on removing the underlying trigger (in this case, malignancy), and immunosuppression. Immunosuppression therapies include systemic glucocorticoids, intravenous immunoglobulin, and plasmapheresis; medications such as mycophenolate mofetil and tacrolimus are options if there is concern for a T-cell mediated process.15
Prion diseases
Prion diseases (spongiform encephalopathy) are a class of neurodegenerative diseases caused by cerebral deposition of misfolded protein and characterized by long incubation periods followed by rapid progression once clinical symptoms present.16 Sporadic CJD (sCJD) is by far the most common prion disease, accounting for 90% of all cases.17
The classic clinical manifestations of sCJD are mental deterioration and myoclonus.18 Most patients are 50 to 70 years old and demonstrate rapidly progressive cognitive impairment and confusion, sometimes with cortical visual disturbances and ataxia. The cognitive syndrome of sCJD is the most commonly reported early symptom (40%), but it can be preceded by mild psychiatric symptoms such as malaise, anxiety, mood changes, and decreased ability to concentrate.19
Sleep disturbances, especially hypersomnia, are also common and may be a presenting sign of sCJD.20 Visual disturbances and oculomotor dysfunction are rarely an early symptom (7%)19 but frequently occur (42%) during the clinical course.21 On neurologic examination, dementia patterns may include apraxia, aphasia, inappropriate jocularity, inability to follow commands, and inattention.22 Involuntary movements can include myoclonus, chorea, dystonia, and tremor. In fact, myoclonus is present in more than 90% of patients with sCJD at some point during their illness.23 Thus, sCJD should be included in the differential diagnosis in any patient presenting with a history of rapidly progressive dementia and myoclonus.
CASE CONTINUED
After ruling out the more common causes of the patient’s worsening altered mental status, diagnostic laboratory testing was pursued for antibody-mediated and paraneoplastic encephalitis and prion disease.
On day 3 of her hospitalization, results from an autoantibody panel were negative, including serum and CSF testing for anti-N-methyl-D-aspartate immunoglobulin G. On this same day, a CSF sample was sent to an outside facility for prion disease biomarker testing. Several days later, blood samples were sent to an outside facility for testing with a paraneoplastic antibody panel. Results would not be available for several weeks.
In the interim, CT of the abdomen, pelvis, and chest was done for possible malignancy. The results were normal.
In the absence of CT findings, positron emission tomography to evaluate for malignancy would have been ideal, given its increased sensitivity and specificity for malignancy of the chest, head, and neck24 but was unavailable at our hospital. Transferring the patient to have the test at a different facility was not possible due to health insurance coverage restrictions.
Therefore, we started empiric treatment for autoimmune and paraneoplastic encephalitis with 5 days of methylprednisolone 1 g, followed by 5 days of 0.2 g/kg of intravenous immunoglobulin, and then 5 days of plasmapheresis.
This treatment has been shown to improve neurologic symptoms (eg, behavior, cognition, speech, memory, seizures) in about half of patients within the first 4 weeks of first-line therapy.25
Her clinical condition continued to deteriorate. Although the paraneoplastic antibody panel testing was still in process at that time, the absence of an identifiable malignancy and lack of improvement with empiric treatment with corticosteroids, intravenous immunoglobulin, and plasmapheresis argued against autoimmune and paraneoplastic encephalitis, increasing our clinical suspicion for prion disease.
NEXT STEP: ADDITIONAL IMAGING
While the diagnostic workup and therapeutic efforts were being pursued, several electroencephalography (EEG) recordings were obtained.
2. What is the most typical EEG finding in a patient with spontaneous CJD?
Sporadic delta-wave activity
Triphasic sharp-wave complexes
K-complexes
Beta-wave activity
Electroencephalography
EEG is an important component in the clinical diagnosis of CJD. A typical pattern of generalized, periodic, biphasic, or triphasic sharp-wave complexes of 1 to 2 Hz is reported in 65% of patients with sCJD.26,27 Periodic sharp waves have also been reported in cases of familial CJD. These EEG changes may not appear until later in the course of the disease, but if nonspecific findings are present on EEG, then frequent serial EEG is recommended.
It is important to recognize that EEG patterns in sCJD are nonspecific and can be observed in other causes of dementia. It should also be noted that variant CJD, a different CJD subtype, does not have the same pattern on EEG as sCJD. Instead, it shows nonspecific slow wave activity without periodic triphasic complexes. Sporadic delta waves are characteristic of physiologic stage N2 and N3 sleep, and K-complexes are characteristic of stage N2 sleep. Beta-wave activity on EEG is characteristic in wakefulness and rapid-eye-movement sleep.28
Magnetic resonance imaging
MRI is a useful diagnostic tool in the context of suspected CJD. In sCJD, T2-weighted MRI with fluid-attenuated inversion recovery (FLAIR) will often show hyperintensity of the putamen and the head of the caudate (sensitivity 67%, specific ity 93%), although numerous etiologies, including toxic, metabolic, hypoxic, and vascular, can cause hyperintensity within the basal ganglia.29 In 90% of cases of variant CJD, T2-weighted MRI demonstrates hyperintensity of the posterior (pulvinar sign) (sensitivity 92%, specificity 95%) and dorsomedial thalamus (hockey-stick sign).23 Table 1 lists the sensitivity and specificity of diagnostic tests for CJD.26,27,30–34
Diagnostic tests for Creutzfeldt-Jakob disease: Sensitivity and specificity
In both sCJD and variant CJD, diffusion-weighted imaging on MRI in particular has been shown to detect disease with high sensitivity (96%) and specificity (93%). Cortical diffusion restriction (cortical ribboning) is a characteristic feature of sCJD but can also be seen in the acute phase of viral encephalitis and focal status epilepticus. Further, cortical ribboning decreases in late stages of sCJD.35
That said, hyperintensity that is more pronounced on diffusion-weighted imaging than on FLAIR has been determined to be crucial when differentiating sCJD from non-prion causes of rapidly progressive dementia, as was seen in all 48 cases in a cohort of sCJD patients.32 Based on these findings, neuroradiologists at the University of California, San Francisco, proposed MRI criteria for the diagnosis of definite CJD, including diffusion-weighted imaging and FLAIR hyperintensity within the cortex (> 1 gyrus) and striatum or only in the cortex (> 3 gyri).32
Although CT is often one of the first tests ordered in the emergency department for assessment of altered mental status, it is not associated with distinct findings for CJD.23
CASE CONTINUED
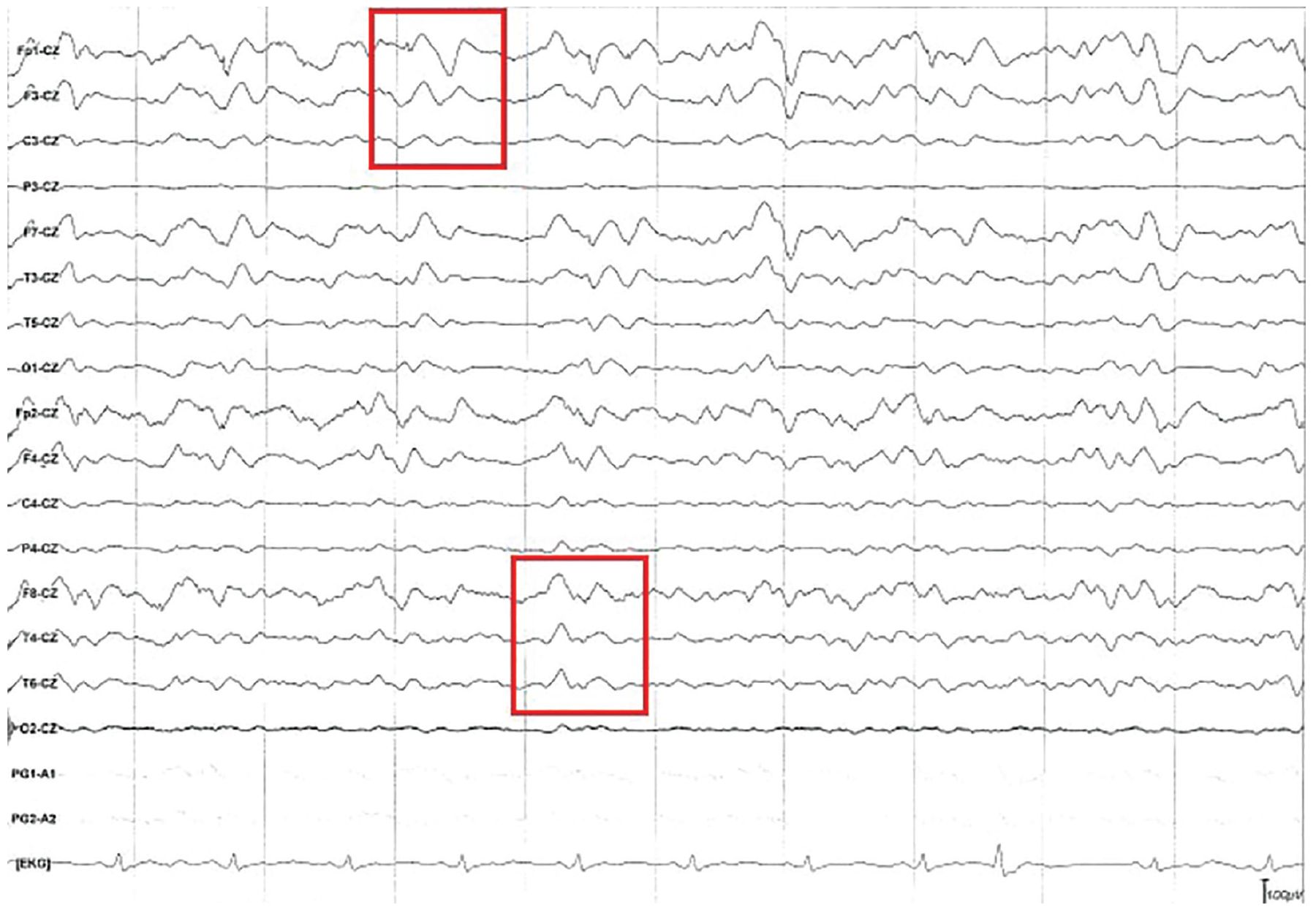
To investigate acute cognitive decline with myoclonus, EEG performed following admission and again on hospital day 11 showed diffuse cortical slowing, with triphasic wave morphology present throughout the tracing (Figure 2). No seizures were observed during the studies, effectively ruling out status epilepticus.

Electroencephalography showed continuous triphasic waves (red boxes) and diffuse cortical slowing. Diffuse slowing is seen throughout the recording, as the background frequency consists mostly of theta waves (frequency 4–7 Hz) despite provoking maneuvers and the patient not being on sedating medications.
However, after multiple episodes of seizure-like activity and decline in mentation, the patient was upgraded to critical care on day 14. On day 17, EEG again showed diffuse slow waves with triphasic morphology but no seizure activity, despite the presence of posturing movement during the study. The interpreting neurologist noted that the findings on EEG might be associated with CJD. Repeat MRI on day 20 showed resolution of cortical diffusion restriction and continued paucity of thalamic hyperintensity. These findings were also suspicious for sCJD, as the cortical ribboning seen on MRI in sCJD often fades late in the disease course.35
On day 18, due to our patient’s continued decline in respiratory and neurologic function (her Glasgow Coma Scale score had dropped to 8), she was intubated and started on enteral tube feedings. This decision was based on doubts about the diagnosis (although there was growing concern for prion disease at this point) and the next of kin’s wishes that full medical interventions be pursued until additional family could visit the patient. In cases of likely or definite diagnosis of CJD and other end-of-life scenarios, intubation and enteral feeding are not recommended.
On day 20, the paraneoplastic panel returned negative results for all tested antibodies, including anti-Ma and anti-Hu antibodies. Carotid artery angiography to assess for vasculitis was also negative. The patient experienced several seizure-like events, and decorticate posturing was noted on day 22.
At this point, diagnostic testing had been either negative or yielded only nonspecific findings, and attempted treatments had failed to stall or improve the patient’s neurologic decline. The only pending test result was for CSF prion disease biomarkers, which had been sent out on day 3.
3. Which of the following is the most helpful CSF test to order if you suspect prion disease?
Myelin basic protein
Oligoclonal bands
Amyloid beta 1-42
14-3-3 protein
Real-time quaking-induced conversion (RT-QuIC)
Neuron-specific enolase
WHY TEST FOR PRION DISEASE?
CSF analysis can provide additional data if the diagnosis of CJD is uncertain. RT-QuIC monitors for formation of amyloid in real time after adding disease-associated prion protein (PrPSc) from the patient (if present) to recombinant prion protein. The mixture is shaken vigorously, exploiting the ability of PrPSc to induce misfolding of recombinant prion protein, forming aggregates. The formation of the aggregates is monitored in real time using a fluorescent dye, thioflavin T.36 Currently, the National Prion Disease Pathology Surveillance Center at Case Western Reserve University in Cleveland, OH, is the only facility in the United States that performs the RT-QuIC assay.37
The 14-3-3 protein is believed to be a marker of massive neuronal disruption and leakage of brain proteins into the CSF.38 The protein biomarker total tau (t-tau), another marker of neuronal death, has been found to be elevated in CSF in patients with sCJD, with 1 study showing it to be a more specific but less sensitive test than the 14-3-3 protein assay.39
The American Academy of Neurology previously recommended CSF testing for the 14-3-3 protein to decrease uncertainty of diagnosis in patients with rapidly progressive dementia and strong suggestion of sCJD.40 However, RT-QuIC has been shown to be a much more powerful diagnostic assay. The American Academy of Neurology has not updated its recommendation for testing for sCJD since 2012, around the same time that studies showing the favorable test characterstics of RT-QuIC began to be published. A retrospective analysis of 111 pathologically confirmed sCJD cases found that RT-QuIC had superior sensitivity and specificity in the diagnosis of sCJD when compared with 14-3-3 protein or t-tau (Table 1).34 A prospective analysis of these data showed similar results.34
Making a probable diagnosis
As such, a positive RT-QuIC assay is a highly weighted component of the scoring systems used to make the probable diagnosis of sCJD (Table 2).41 However, RT-QuIC can be less sensitive in some molecular subtypes of sCJD, so a negative test does not necessarily rule out the disease. In those cases, 14-3-3 protein results, clinical presentation, and characteristic findings on MRI and EEG can aid the diagnosis.34
Criteria for probable diagnosis of sporadic CJD
It should be noted that the most recent World Health Organization guidelines for diagnosis of CJD (released in 2003)23 do not take RT-QuIC into account, and thus it may be considered out of date.
High levels of myelin basic protein and oligoclonal Ig G bands are CSF findings useful to diagnose demyelinating disorders such as multiple sclerosis.42 Amyloid beta (along with t-tau, p-tau, and tau-tau ratio)9 is a protein essential to the pathogenesis of Alzheimer disease, implicated in free radical-induced oxidative stress.43 Neuron-specific enolase is a marker that has great utility in the evaluation of both small cell and non-small cell lung cancers, stroke and brain injury, neuroendocrine tumors, and neuroblastoma.44
CASE CONTINUED
On day 26, the off-site CSF analysis returned positive results for RT-QuIC, 14-3-3 protein, and t-tau protein. In combination, these results are nearly 100% specific for sCJD and make other causes of dementia, such as Alzheimer disease, frontotemporal dementia, or Lewy body dementia, unlikely. The poor prognosis of CJD was discussed with the patient’s sister, but further action was deferred.
The patient was noted to have continued posturing and no longer withdrew from painful stimuli or tracked objects. When the patient’s brother arrived on day 31, the family requested palliative care only. The patient was extubated and prescribed midazolam and fentanyl. She exhibited labored breathing with substernal retractions and died on hospital day 37.
EPIDEMIOLOGY OF PRION DISEASES
As noted earlier, prion diseases are neurodegenerative diseases with long incubation periods but with rapid progression once symptoms emerge. There are 5 recognized prion diseases: kuru, CJD, variant CJD, Gerstmann-Straussler-Scheinker syndrome, and fatal familial insomnia. Of these, CJD accounts for more than 90% of prion disease cases.17 However, the low incidence of CJD—about 1 case per 1 million individuals per year16—can present diagnostic challenges to practitioners unfamiliar with the disease.
Sporadic, familial, iatrogenic, and variant forms of CJD are all recognized (variant CJD is sometimes categorized separately because of its distinct clinical and pathological findings).45 The vast majority of CJD cases (85% to 95%) are sporadic. Familial CJD accounts for 5% to 15% of cases but is much less common, accounting for fewer than 1 case per 10 million people.18,23 However, a single autosomal dominant trait (PRNP E200K–129M) accounts for 70% of familial CJD cases, which are clustered among populations in Chile, Italy, Japan, and Slovakia, and in Jews from Libya.46 Variant CJD is the disease type transmitted from bovine spongiform encephalopathy. As of February 2020, only 235 cases of variant CJD had been reported since 1980.47
What are the risk factors for CJD?
Several studies have attempted to identify risk factors for sCJD. A review of 3 case-control studies published in 1996 showed that a family history of CJD (odds ratio [OR] 19.1) and a medical history of psychosis (OR 9.9) were the only factors significantly associated with the disease.48 A study conducted in Australia found that living or working on a farm for more than 10 years was associated with a significantly increased risk for sCJD (OR 2.61, 95% CI 1.34–3.41).49 A systematic review published in 2017 reported, based on very low-quality evidence, that sCJD was associated with heart (OR 1.96) and vascular (OR 2.13) surgery.50 However, the low incidence of the disease makes it difficult to assess predisposing factors.
PATHOLOGY DRIVES PRECAUTIONS
Classic neuropathologic findings in CJD are marked neuronal loss, spongiform change, and astrogliosis. However, immunohistochemical staining for prion protein is considered the technical standard for diagnosing CJD (Figure 3).51
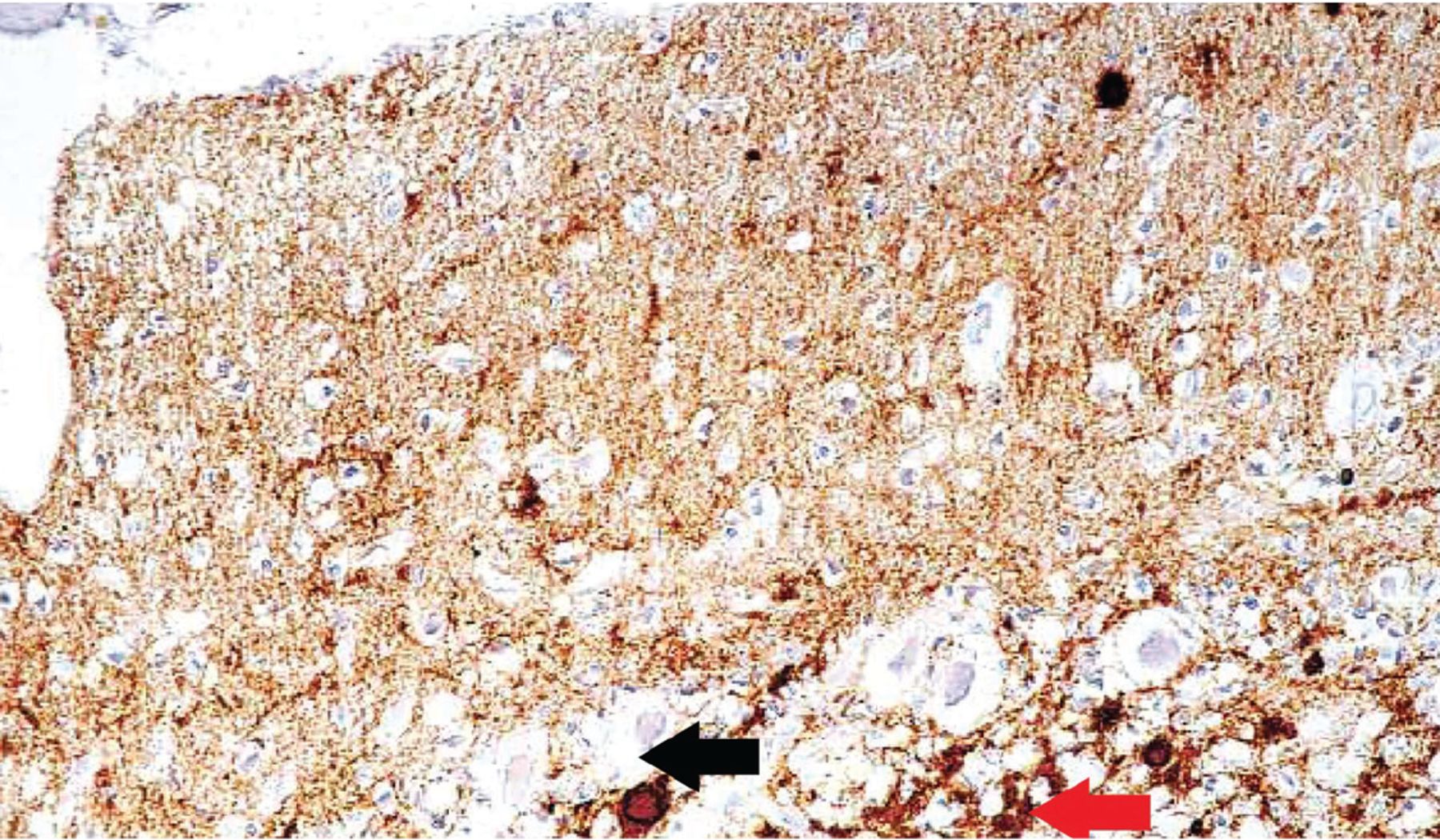
Immunohistochemical staining shows fine prion protein deposits in the molecular layer, coarser deposits in the granular layer, and plaques in both layers of the cerebellum (magnification × 600). Fine deposits in the upper portion (molecular layer) of the image appear as numerous dark deposits. The red arrow points to coarse deposits in the granular layer, and the black arrow points to plaque.
From Kovács GG, Head MW, Hegyi I, et al. Immunohistochemistry for the prion protein: comparison of different monoclonal antibodies in human prion disease subtypes. Brain Pathol 2002; 12(1):1–11. doi:10.1111/j.1750-3639.2002.tb00417.x. Copyright John Wiley and Sons, Inc. Reprinted with permission.
4. What precautions are required if CJD is suspected or diagnosed?
Strict isolation with hazardous material suit
Contact precautions with gloves and gowns
Droplet precautions
Airborne precautions
Strict universal precautions, special attention to instrument-, body fluid-, and tissue-handling, and transport
Prion diseases are transmitted through contaminated instruments and infected tissues, with different tissues being categorized as having high or low infectivity. High-infectivity tissues include brain, spinal cord, eye tissues, spinal ganglia, and trigeminal ganglia. Low-infectivity tissues include CSF, peripheral nerves, blood, kidney, liver, lung, lymph nodes and spleen, and placenta.52 There have been only 4 cases of variant CJD transmitted via blood transfusion.53 No person-to-person transmission has been reported through usual contact. If a patient is suspected to have CJD, the following measures should be taken to prevent iatrogenic or nosocomial exposure to prion disease:
Screen donor sources of dura and cornea
Label all reusable instruments that have contacted low-or high-infectivity tissue as “biohazard,” place them in a robust, leak-proof container, and transport them to sterilization as soon as possible after use
Incinerate all disposable instruments and treat heat-resistant instruments with sodium hydroxide
Treat CSF as if it were highly infective tissue
Take World Health Organization precautions for high-and low-infectivity tissues from patients with known or suspected CJD.23
TREATMENT AND PROGNOSIS
Unfortunately, there is no curative treatment for the underlying disease process of CJD. Attempts at treatment of myoclonus using clonazepam or valproate may be helpful for palliation.23
Prognostically, CJD is characterized by a rapidly deteriorating course. Death usually occurs within 1 to 2 years of symptom onset, most often from aspiration pneumonia.22,23 However, the time from presentation to death can vary among the subtypes of CJD. Based on a systematic review of more than 9,000 patients, sCJD and familial CJD have the most rapid clinical deterioration (median mortality 6 months), iatrogenic CJD has a slightly longer course (median mortality 9 months), and variant CJD and inherited prion disease have the longest course (median mortality 14 months).23
Within sCJD, there are various molecular subtypes characterized by the presence of a valine or methionine allele at codon 129 of the prion protein gene, as well as the type of PrPSc (type 1 vs type 2), that can also affect the prognosis. For example, homozygosity for methionine at codon 129 and expression of PrPSc type 1 is the most common subtype and has the shortest duration from symptom onset to death (mean 3.9 months). Meanwhile, methionine-valine heterozygosity and expression of PrPSc type 2 has a more prolonged course (mean 17.1 months).54
Genetic testing for prion protein gene allelic mutations is available for patients with a family history of CJD. A diagnosis of familial CJD can be confirmed with a recognized prion protein mutation (of which there are at least 41 from unrelated families) and a definite or probable transmissible spongiform encephalopathy in a first-degree relative.
The National CJD Research & Surveillance Unit (based at the University of Edinburgh, Scotland, UK) recommends genetic analysis for prion protein codon mutations to exclude the possibility of genetic disease.55 All patients who die with suspected or probable sporadic or variant CJD should have brain tissue frozen and sectioned postmortem to confirm the presence of PrPSc and make the definitive diagnosis.
TAKE-HOME MESSAGES
There are many possible infectious and neurodegenerative causes of dementia, making the differential diagnosis broad. Given our patient’s lack of risk factors for CJD (eg, no family history of psychosis or CJD), it was low on the initial list of diagnostic considerations. However, through a systematic approach and diagnostic workup, the relatively common causes were quickly ruled out, thus increasing suspicion for CJD.
Practitioners can rely on the patient’s medical history and a thorough neuropsychiatric assessment to guide clinical suspicions. If support is lacking for more common diagnoses, the clinician can reach a probable diagnosis of sCJD through CSF assay and commonly ordered imaging studies.
Although prompt diagnosis does not alter the prognosis, it does provide benefits in 2 important ways. First and foremost, it helps to prevent the family from developing unrealistic expectations of patient recovery, informs decision-making on goals of care, and allows them to appropriately grieve for their loved one. Second, it serves as an educational opportunity for all healthcare professionals involved, encouraging them to widen their differential diagnosis, consider uncommon investigations, and communicate complex ideas to both colleagues and patients.
DISCLOSURES
The authors report no relevant financial relationships which, in the context of their contributions, could be perceived as a potential conflict of interest.
- Copyright © 2021 The Cleveland Clinic Foundation. All Rights Reserved.
REFERENCES
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- DIFFERENTIAL DIAGNOSIS: DISEASES, CONDITIONS TO RULE OUT
- CASE CONTINUED
- AUTOANTIBODY ENCEPHALITIS: WHAT TO LOOK FOR
- CASE CONTINUED
- NEXT STEP: ADDITIONAL IMAGING
- CASE CONTINUED
- WHY TEST FOR PRION DISEASE?
- CASE CONTINUED
- EPIDEMIOLOGY OF PRION DISEASES
- PATHOLOGY DRIVES PRECAUTIONS
- TREATMENT AND PROGNOSIS
- TAKE-HOME MESSAGES
- DISCLOSURES
- REFERENCES
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.