


ABSTRACT
Potentially deadly drug rashes include Stevens-Johnson syndrome/toxic epidermal necrolysis, drug reaction with eosinophilia and systemic symptoms, acute generalized exanthematous pustulosis, and drug-induced vasculitis. Differentiating them can be a challenge. Factors to consider include timing of rash to drug exposure, rash distribution and clinical appearance, and the presence of systemic features such as mucosal involvement, organ failure, or eosinophilia. Various scoring systems aid in the diagnosis, but skin biopsy is the gold standard. Prompt identification and withdrawal of the suspected offending agent are the crucial first steps in management.
Differentiating severe drug rashes involves consideration of timing of drug exposure, clinical appearance of the rash, presence of systemic features, and often skin biopsy.
Early recognition and immediate withdrawal of offending agents is critical to minimize debilitating and potentially life-threatening consequences of severe drug rashes.
Pharmacologic treatment depends on the rash and is controversial, with inconsistent published outcomes. A multidisciplinary approach with supportive measures is key to reducing morbidity and mortality.
Adverse drug reactions (ADRs) are the fifth leading cause of death among all diseases and account for 5% to 10% of hospitalizations worldwide.1 They remain a challenge in modern healthcare, particularly with increasing complexity of comorbidities and therapeutics.
By definition, ADRs are unintended harmful events attributed to the use of medicines in clinical practice. They are associated with prolonged hospital courses, increased rates of readmission and costs of patient care, and death, and 30% to 45% involve the skin.1 Risk factors include female sex, older age, higher numbers of drugs, immunocompromised status, and autoimmune disorders.1
Identifying the type of drug rash is a challenge. Clinicians are familiar with the clinical features of the 2 most common drug-induced cutaneous reactions, morbilliform drug rash and urticarial rash2:
Morbilliform drug rash, also called exanthematous or maculopapular drug eruption, is the most common, classically presenting with an erythematous maculopapular rash 1 to 2 weeks after a drug exposure
Urticarial rash, the second most common, presents as annular, pruritic, migratory plaques usually within hours of initial drug exposure.2
Cutaneous reaction rates are highest with penicillins, sulfonamides, anticonvulsants, and nonsteroidal anti-inflammatory drugs (NSAIDs), and NSAIDs and salicylates are more commonly associated with urticarial than with morbilliform drug rash.3 Severe drug rashes are less common but can be life-threatening. Early recognition and prompt immediate withdrawal of the suspected drug is crucial.
This article reviews the distinguishing features of 4 severe drug rashes, summarized in Table 1: Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN), drug reaction with eosinophilia and systemic symptoms (DRESS), acute generalized exanthematous pustulosis (AGEP), and drug-induced vasculitis.
Characteristics of selected high-risk drug rashesa
STEVENS-JOHNSON SYNDROME/TOXIC EPIDERMAL NECROLYSIS
SJS and TEN are overlapping conditions characterized by mucocutaneous reactions with epidermal necrosis and detachment. The conditions are classified into 3 categories of severity based on the percentage of body surface involved:
SJS: lesion area is less than 10%
SJS/TEN overlap: lesion area is 10% to 30%
TEN: lesion area is greater than 30%.
The estimated overall incidence of SJS/TEN in Europe and the United States is up to 6 cases per million person-years. The rates are higher in adults, females, and people of Asian or Black ethnicity. The most common inciting drugs are allopurinol, antibiotics (particularly sulfonamide antibiotics), antiepileptics, and NSAIDs.4 Immune checkpoint inhibitors, which are increasingly prescribed for malignancy, are associated with severe cutaneous drug eruptions, including SJS/TEN.5
Symptom onset after 1 to 3 weeks
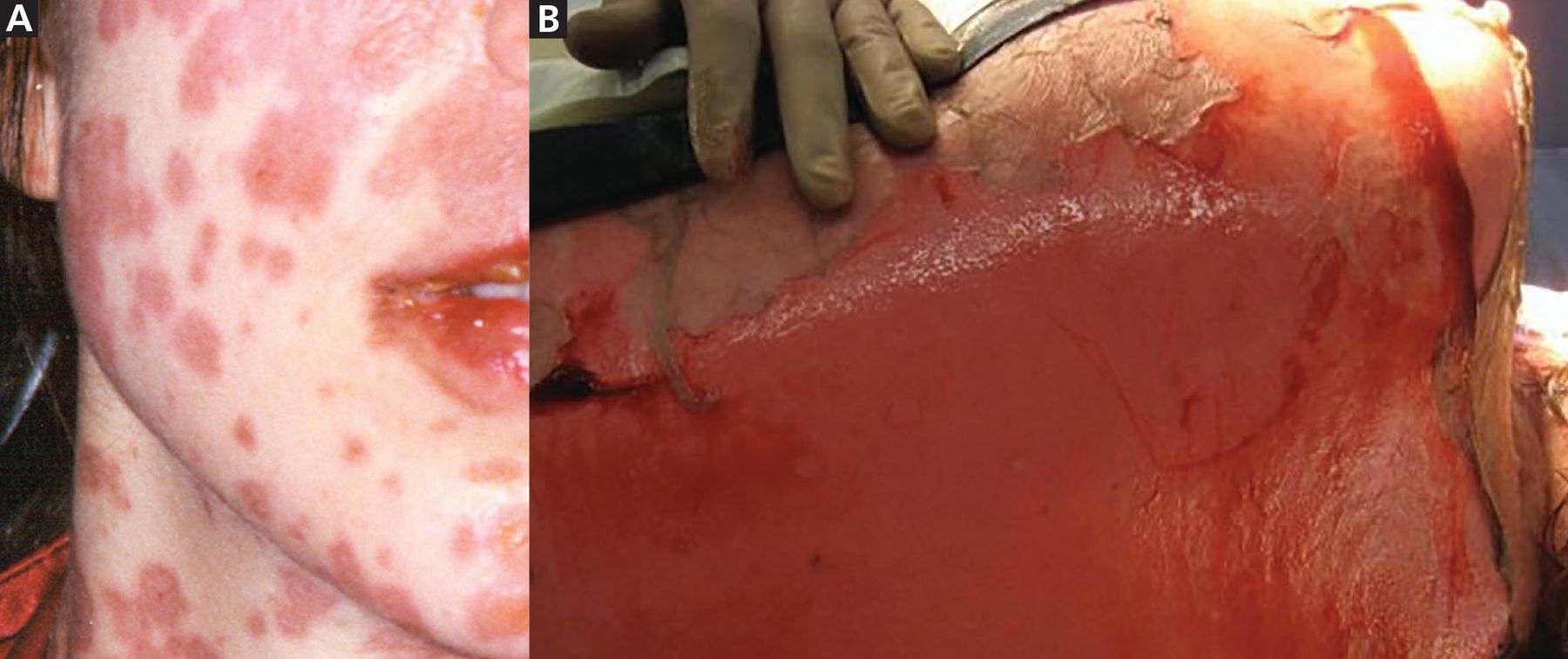
Rash onset is usually 1 to 3 weeks after drug introduction. Typically, lesions appear first on the face and thorax before spreading symmetrically. They start as macules and target-like lesions with erythema and dark necrotic centers and develop into vesicles, erosions, or ulcerations with epidermal detachment. They often have a positive Nikolsky sign, ie, where traction pressure causes epidermal shearing and erosion (Figure 1).6

(A) Macules and target-like lesions with erythema and dark necrotic centers in Stevens-Johnson syndrome. (B) Positive Nikolsky sign with epidermal shearing in Stevens-Johnson syndrome.
Systemic manifestations
Systemic manifestations are common and include flulike symptoms, fever, lymphadenopathy, and mucosal involvement (conjunctival, oropharyngeal, esophageal, and genital). Mucosal involvement occurs in up to 90% of patients, and mouth ulcers, grittiness in the eyes, odynophagia, and dysuria are common.4,6
The most clinically significant elements of mucosal involvement are the sequelae of mucosal ulceration that result in scarring and stricture, which affect several organ systems—namely, the cornea, urethra, esophagus, and pulmonary tract. Severe complications of SJS/TEN include respiratory failure, shock, functional volume depletion, and infections. The average mortality rate is 1% to 5% in SJS and 25% to 35% in TEN.4,7
Diagnosis
The diagnosis of SJS/TEN is based on a history of drug exposure along with clinical evidence of classic mucocutaneous lesions. The gold standard for diagnosis is skin biopsy with routine histopathology and direct immunofluorescence studies. Biopsy can be helpful even in early stages if the diagnosis is uncertain, but it is more definitive in later stages with the hallmark manifestations of full-thickness necrosis and subepidermal detachment. Biopsy at this later stage helps exclude diagnoses that mimic SJS/TEN. These include staphylococcal scaled-skin syndrome and other generalized rashes with blisters, such as exfoliative erythroderma, bullous pemphigoid, pemphigus vulgaris, and linear immunoglobulin A dermatosis.8
Supportive care and prompt referrals are essential
The first and most important step in management of a patient with SJS/TEN is immediate identification and withdrawal of the suspected offending medications. Prompt withdrawal of the causative agent before erosions and blisters develop significantly reduces the risk of death.9 The SCORTEN tool (Severity-of-Illness Score for Toxic Epidermal Necrolysis) includes prognostic indicators such as heart rate, age, and renal function and can be used to determine a patient’s risk of death with SJS/TEN (Table 2).4,10
Severity-of-illness Score for Toxic Epidermal Necrolysis (SCORTEN)
The mainstay of treatment is supportive care: intravenous fluids, electrolyte replacement, nutritional support, pain control, and prevention of infection. Interval skin cultures and blood cultures can aid in early detection and treatment of superinfection.11 Prompt referral to burn units and specialists (eg, ophthalmology, urology) based on organ involvement is indicated.
Treatment with corticosteroids is controversial, but intravenous immunoglobulin (IVIG) therapy alone or in combination with corticosteroids has shown varying degrees of success.12,13 Other options include plasmapheresis, immunosuppressive agents (cyclosporin, cyclophosphamide, thalidomide), or various combinations of these options and any of the above treatments.14 Prophylactic systemic antibiotics should be avoided unless a workup for infection raises concern for bacterial superinfection.15
DRUG REACTION WITH EOSINOPHILIA AND SYSTEMIC SYMPTOMS
DRESS is a delayed-onset multiorgan reaction. The onset is usually 2 to 6 weeks after initiation of medication, although a rash can be seen earlier with medications such as antibiotics.16 The incidence is 1 in 1,000 to 10,000 drug exposures, and it is responsible for about 18% of inpatient adverse drug reactions that affect the skin.17,18 The most common offending drugs include antiepileptics (carbamazepine, phenytoin, lamotrigine, phenobarbital), allopurinol, sulfonamides (sulfasalazine, dapsone, trimethoprim-sulfamethoxazole), minocycline, vancomycin, and antituberculosis agents (isoniazid, rifampicin, ethambutol, pyrazinamide).19
Fever, rash, facial edema, eosinophilia
DRESS often starts with fever and a rash, characterized as a nonspecific severe pruritic skin eruption affecting more than 50% of the body surface area. Patients often develop severe facial edema that is central with periorbital sparing. The rash is usually maculopapular, but lesions are polymorphous and can present as plaques, blisters, target-like lesions, urticaria, exfoliation, eczema, or, rarely, lichenoid eruptions (Figure 2).

(A) A maculopapular rash in a drug reaction with eosinophilia and systemic symptoms. (B) Lesions may also present as plaques, blisters, or target-like lesions.
In addition to rash and fever, other manifestations may include lymphadenopathy, hematologic abnormalities, and internal organ involvement (most commonly liver, kidney, lung, and cardiac injury). Up to 95% of patients with DRESS have eosinophilia.20 With a prolonged clinical course, sequential reactivation of various human herpesviruses (particularly type 6 and type 7) and, less frequently, Epstein-Barr virus and cytomegalovirus infections, may be seen.21
The course can wax and wane with multiple flares. The average mortality rate is 4% to 10% from multiorgan failure (most commonly hepatic necrosis),22 with long-term complications that include exfoliative dermatitis,23 acute necrotizing eosinophilic myocarditis, and autoimmune sequelae such as thyroid disease, vitiligo, alopecia areata, lupus erythematosus, autoimmune hemolytic anemia, and fulminant type 1 diabetes mellitus.22–25
RegiSCAR: Resource for diagnostic criteria
The clinical presentation of rash, eosinophilia, and internal organ involvement should prompt an evaluation for possible DRESS. The RegiSCAR criteria (Registry of Severe Cutaneous Adverse Reactions) are the most detailed and frequently used diagnostic criteria (Table 3).21 Follow-up bloodwork should be obtained based on suspected organ involvement. Histopathology for DRESS is nonspecific and includes spongiosis, basal vacuolization, necrotic keratinocytes, dermal-epidermal infiltrates, dermal edema, and perivascular infiltrates of lymphocytes with or without eosinophils.7,21
Registry of Severe Cutaneous Adverse Reactions diagnostic criteria for drug reaction with eosinophilia and systemic symptoms
Identifying the causative agent may be a challenge because of the delayed presentation after drug exposure. Lymphocyte transformation testing is the most reliable in vitro method to confirm the causative drug, and is particularly useful for confirming anticonvulsant and antituberculosis therapies. It assesses activation of drug-specific T cells with 73% sensitivity and 82% specificity, but must be performed 2 to 6 months after the acute phase.21,26 In vivo skin testing, particularly patch testing and delayed intradermal testing, can also be useful in identifying the causative drug.21,26
Multidisciplinary management
Management of DRESS requires a multidisciplinary approach based on organ involvement and severity. If the patient has mild disease with a modestly elevated transaminase (< 3 times upper limit of normal), treatment is symptomatic with topical corticosteroids.27 Moderate- to high-dose systemic corticosteroid therapy is the treatment of choice for severe disease. For corticosteroid-resistant patients, IVIG and Janus kinase inhibition have shown some success. Other alternatives include immunosuppressive agents (cyclophosphamide, cyclosporine, interferons, mycophenolate mofetil, rituximab), antivirals, and plasmapheresis. Antibiotics and antipyretics should be avoided unless there is definite evidence of infection.21
ACUTE GENERALIZED EXANTHEMATOUS PUSTULOSIS
AGEP is a severe rapid cutaneous pustular reaction that usually occurs within 48 hours of drug exposure. Its incidence is 1 to 5 cases per million person-years, and common causative drugs are antibiotics, antimycotics, hydroxychloroquine, and diltiazem.28
Abrupt presentation
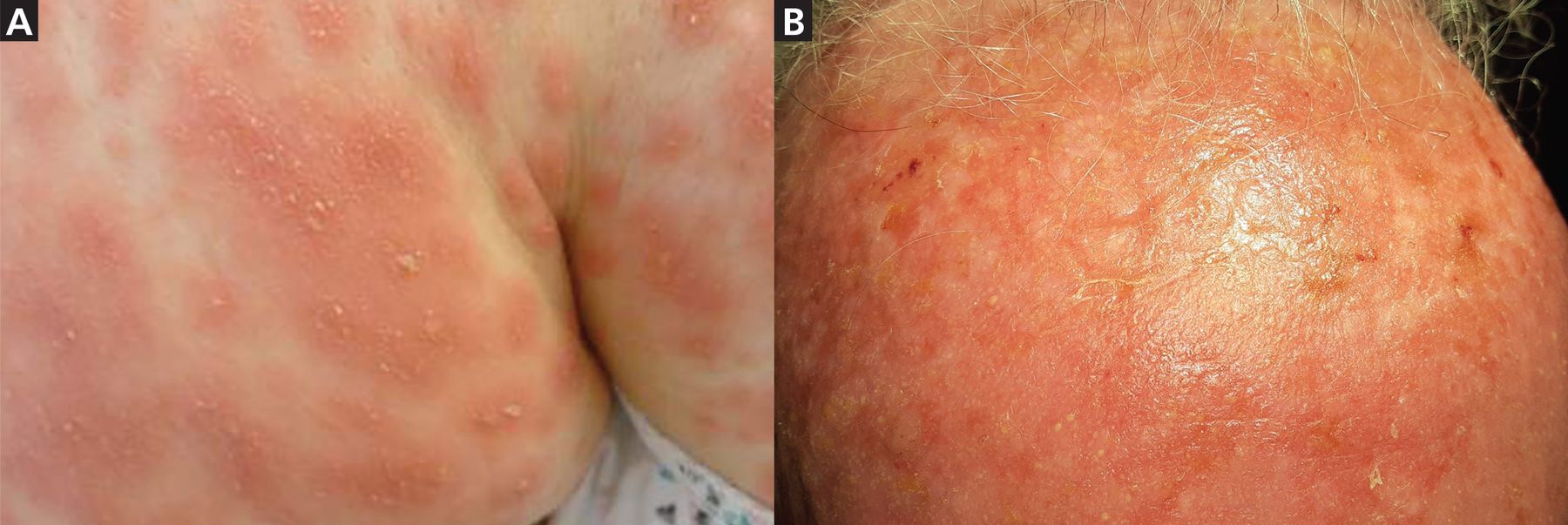
AGEP presents abruptly with hundreds of pinhead-size pustules on a background of diffuse edematous erythema (Figure 3). It usually starts in the intertriginous folds or on the face, or both, and later spreads to the trunk and extremities. Lesions can cause burning and pruritus, and mucosal involvement is rare.

(A) Pustules and diffuse edematous erythema in a patient with acute generalized exanthematous pustulosis affecting intertriginous folds and, (B) a patient’s forehead.
The rash is associated with fever, leukocytosis (predominantly neutrophilia), elevated C-reactive protein level, and in 20% of patients, multiorgan involvement.29 Pustules resolve spontaneously within a few weeks and are followed by postpustular pinpoint desquamation described as collarette-shaped. The overall mortality rate is less than 5%, mostly from complications such as skin superinfection, multiorgan dysfunction, and disseminated intravascular coagulation.7,29
Dermoscopy enhances early diagnosis
The EuroSCAR diagnostic score (European Study of Severe Cutaneous Adverse Reactions) can be used to define clinical and diagnostic criteria (Table 4).30 Pustules are often difficult to visualize, but dermoscopy with a magnifier and polarized light can enhance early diagnosis with detection of pustules at an early stage. Skin biopsy usually reveals intracorneal, subcorneal, and intraepidermal pustules with papillary dermal edema and infiltrates with neutrophils and eosinophils, occasionally including epidermal changes such as spongiosis with necrotic keratinocytes. When the cause of AGEP is unclear, patch testing after resolution of the symptoms may be an option.29
European Study of Severe Cutaneous Adverse Reactions scoring system for acute generalized exanthematous pustulosisa
Prevention of infection with moist dressings and antiseptic solutions is recommended during the pustular phase. In prolonged cases, topical corticosteroids may help relieve symptoms and decrease duration of hospitalization. Antibiotics should be avoided in the absence of superinfection.29
DRUG-INDUCED VASCULITIS
Drug-induced vasculitis is typically a small-vessel vasculitis related to the immune complex-mediated reaction of the dermal capillaries and venules. Drug-induced vasculitis is usually limited to cutaneous vasculitis and arthralgia but, rarely, it can present as severe multiorgan involvement that can mimic primary systemic vasculitis.31 Drug-induced vasculitis typically presents 1 to 3 weeks after drug initiation and is usually self-limited. The most common causative drugs are antibiotics, sulfonamides, diuretics, allopurinol, NSAIDs, amiodarone, beta-blockers, selective serotonin reuptake inhibitors, and metformin.32
The usual presentation is nonblanching palpable petechiae and purpura (Figure 4). The rash is commonly bilateral on dependent areas of the body and sometimes develops into hemorrhagic vesicles and bullae, pustules, nodules, crusted ulcers, or livedo reticularis. Koebnerization, the appearance of lesions at areas of trauma, is uncommon, but reverse koebnerization has been described with the disappearance of the lesions with pressure bandaging following the skin biopsy.32

Rash associated with drug-induced vasculitis. Bilateral presentation on dependent areas of the body is common.
Approximately 30% of patients present with extracutaneous involvement such as arthralgias or renal, gastrointestinal, pulmonary, or neurologic symptoms.32 The mortality rate, about 2%, is usually related to systemic involvement.32
Consider alternative causes
Diagnosis of a drug-induced vasculitis should be guided by the clinical presentation with consideration of alternative causes of systemic vasculitis. A reasonable workup includes basic laboratory testing, infectious serologies (hepatitis B and C, human immunodeficiency virus), serum protein electrophoresis, direct immunofluorescence studies with immunoglobulins (IgG, IgA, IgM), antinuclear antibodies, rheumatoid factor, serum complement levels, antineutrophil cytoplasmic antibodies, and cryoglobulins. Definitive diagnosis can be confirmed with skin biopsy that typically shows any of the following:
Evidence of neutrophilic infiltration within and around the vessel wall with the signs of “clear dust” or leukocytoclasia (disintegration of neutrophil nuclei into fragments)
Fibrinoid necrosis or fibrin deposition within and around the vessel wall
Signs of damage to the vessel wall and surrounding tissue such as damaged endothelial cells or extravasated red blood cells.
When drug-induced vasculitis is suspected, the causative agent should be discontinued immediately. In most cases, the condition is self-limited and responds to supportive care and symptomatic relief including rest, elevation if a dependent extremity is affected, and use of compression stockings. In severe cases, corticosteroids usually bring a rapid response. Other options are colchicine, dapsone, hydroxychloroquine, and NSAIDs. In patients with underlying systemic vasculitis, immunosuppressive medications (azathioprine, methotrexate, mycophenolate mofetil), biologics, or plasma exchange can be considered.33
GENERAL APPROACH: IDENTIFY, CONFIRM, GIVE SUPPORTIVE CARE
The most important clues for identifying and differentiating among deadly drug rashes are in the history, timing of exposure, and the bedside physical examination. While there is overlap, severe drug rashes have distinguishing features and characteristics, reviewed in Table 1.
Generally, when a severe drug rash is suspected, immediate identification and withdrawal of the suspected offending medication is indicated. To aid and support the diagnosis, especially in cases of uncertainty, a definitive diagnosis is often confirmed with skin biopsy. Because of the potential for life-threatening complications and sequelae, management starts immediately with supportive measures: intravenous maintenance fluid, nutritional supplementation, and consultations with burn units or other specialists to minimize long-term sequelae such as ocular, renal, lung, liver, or genitourinary involvement. Specific medical management is complicated and varies depending on the patient and the specific rash.
PREVENTION IS A CHALLENGE
Preventing severe drug rashes is challenging, although gathering a thorough history of past severe adverse drug reactions can help decrease risk of future harm.
There may be a role for human leukocyte antigen testing in prevention of severe adverse drug reactions, as shown in the following 2 examples:
The HLA-B*5801 allele is associated with a markedly elevated risk of allopurinol hypersensitivity syndrome. The prevalence of this allele is highest among persons of Han Chinese, Korean, and Thai descent (7.4%) and African Americans (3.8%).34 The American College of Rheumatology conditionally recommends testing for the HLA-B*5801 allele in these higher risk populations before starting allopurinol.34
The HLA-B*1502 allele is almost exclusively seen in patients with Asian ancestry, and these patients have a higher risk of SJS and DRESS with antiepileptic agents.35 The US Food and Drug Administration recommends screening these at-risk populations before starting carbamazepine, oxcarbazepine, and possibly phenytoin. Future studies are likely to identify other genetic testing that could limit provocation of serious cutaneous adverse drug reactions.
DISCLOSURES
The authors report no relevant financial relationships which, in the context of their contributions, could be perceived as a potential conflict of interest.
- Copyright © 2023 The Cleveland Clinic Foundation. All Rights Reserved.




{kind=link}
{kind=link}
{kind=link}
{kind=link}