


A 72-year-old woman with a history of psoriasis, chronic lower-extremity edema, and hypertension presented to the emergency department with 2 days of a progressive painful blistering rash primarily involving her hands. She had previously experienced infrequent episodes of herpes labialis and symptoms of fatigue and myalgia. She denied any recent travel or contact with chemicals. She was taking losartan 50 mg/day and furosemide 20 mg/day, which had been prescribed 2 months before for hypertension and peripheral edema.
INITIAL EVALUATION AND MANAGEMENT
Her temperature was 98.5°F (36.9°C), heart rate 107 beats per minute, blood pressure 180/83 mm Hg, respiratory rate 18 breaths per minute, oxygen saturation 100% on room air, weight 69 kg (152 lb), and body mass index 26 kg/m2.
Skin lesions were present at several sites. The dorsum of her hands had dozens of clear vesicles 0.5 cm to 2 cm in diameter and bullae (Figure 1), and her palms had red target lesions 0.5 cm to 1 cm (Figure 2). Her thighs had red plaques that had central scale and were bordered by vesicles. Her left lower lip had 2 flaccid vesicles 3 mm to 4 mm. No ocular or anogenital lesions were seen. The lesions were negative for the Nikolsky sign (separation of the epidermis from the dermis).
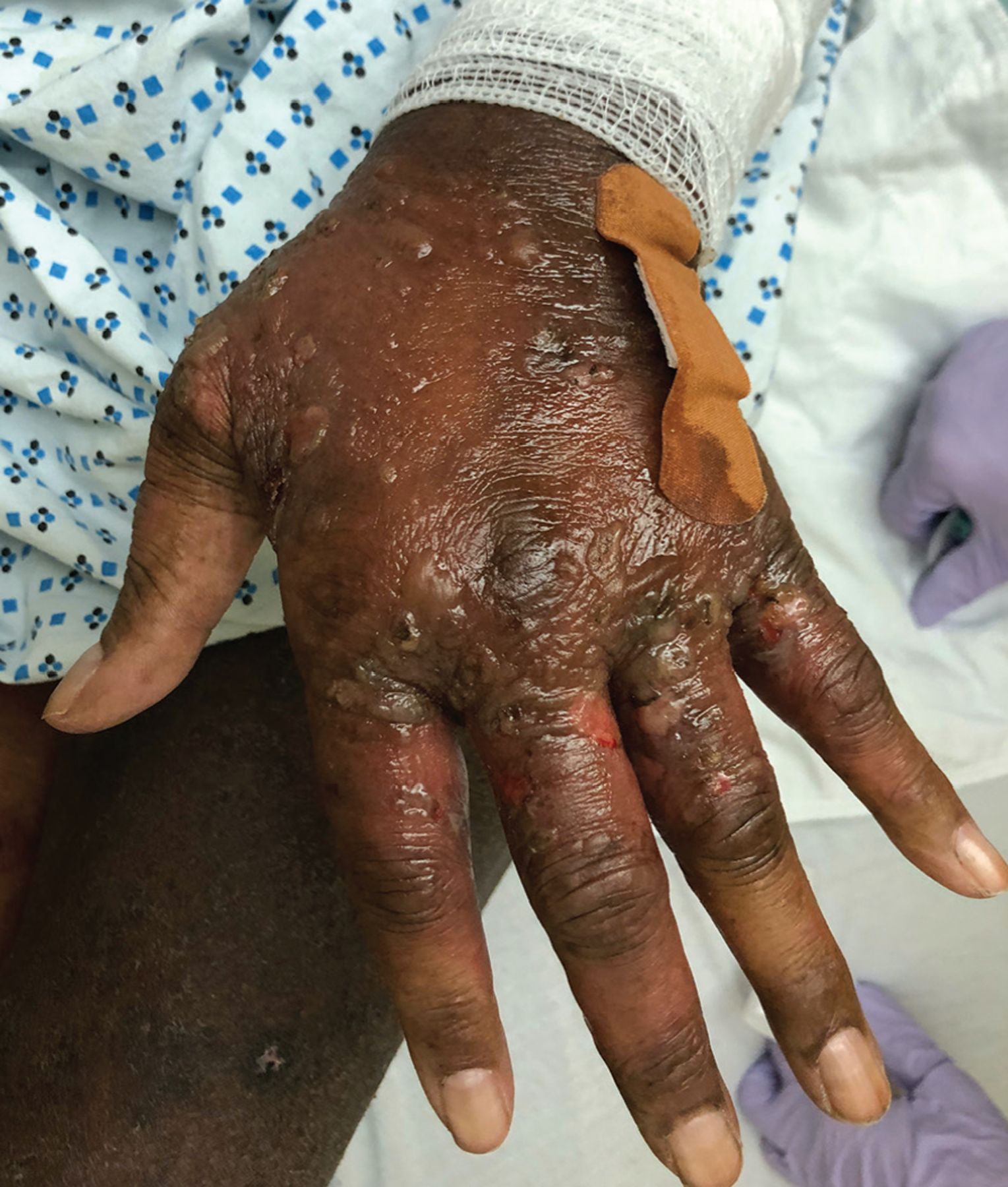
The dorsum of the left hand with tense bullae and erosions.
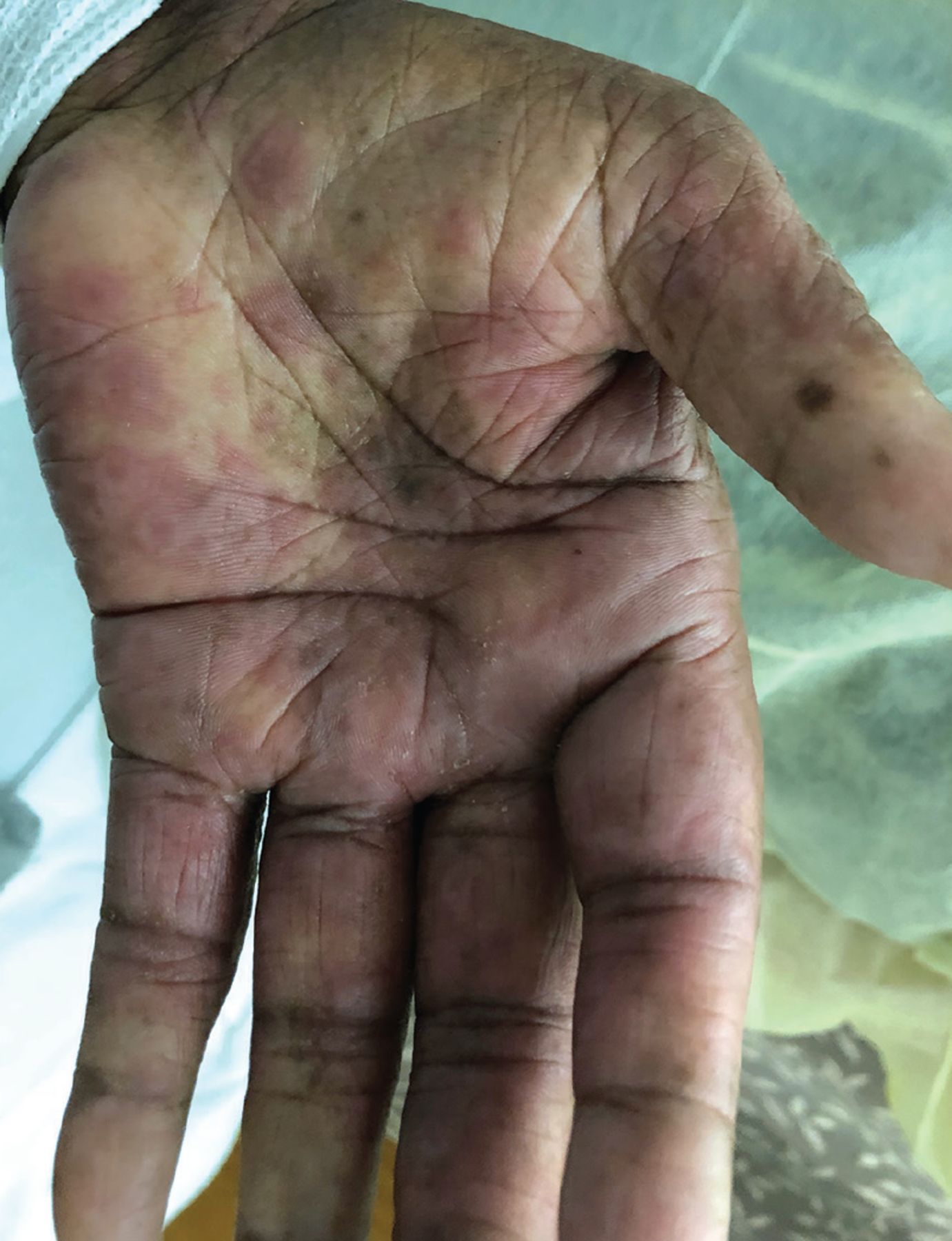
The left palm with red, atypical target lesions.
Her lungs were clear to auscultation. Although her heart rate was elevated, the rhythm was regular, and there were no murmurs, rubs, or gallops. Her abdomen was soft and nontender to palpation. She was alert and oriented, and her behavior was appropriate.
Laboratory testing and histopathology
Laboratory tests taken while she was in the emergency department were notable for the following results:
White blood cell count 12.1 × 109/L (reference range 4.0–10.0 × 109/L)
Eosinophils 2.3% (0.0–7.0%)
Absolute eosinophil count 0.3 × 109/L (0.0–1.0 × 109/L)
Hemoglobin 10.4 g/dL (12.0–18.0 g/dL)
Mean corpuscular volume 78.7 fL (78.0–94.0 fL)
Platelet count 359 × 109/L (140–440 × 109/L)
Erythrocyte sedimentation rate 86 mm/h (0–20 mm/h)
C-reactive protein 80 mg/L (< 10 mg/L).
She was admitted to the hospital and treated empirically with famciclovir for a presumed herpes infection. Further evaluations returned negative results for antibodies to bullous pemphigoid 180 (BP180) and 230 (BP230) and collagen type VII in the serum. Polymerase chain reaction testing was negative for herpes simplex virus (HSV) 1 and 2, varicella, and enterovirus using viral skin swabs from her left hand. Viral swabs from the lip lesions were negative for HSV.
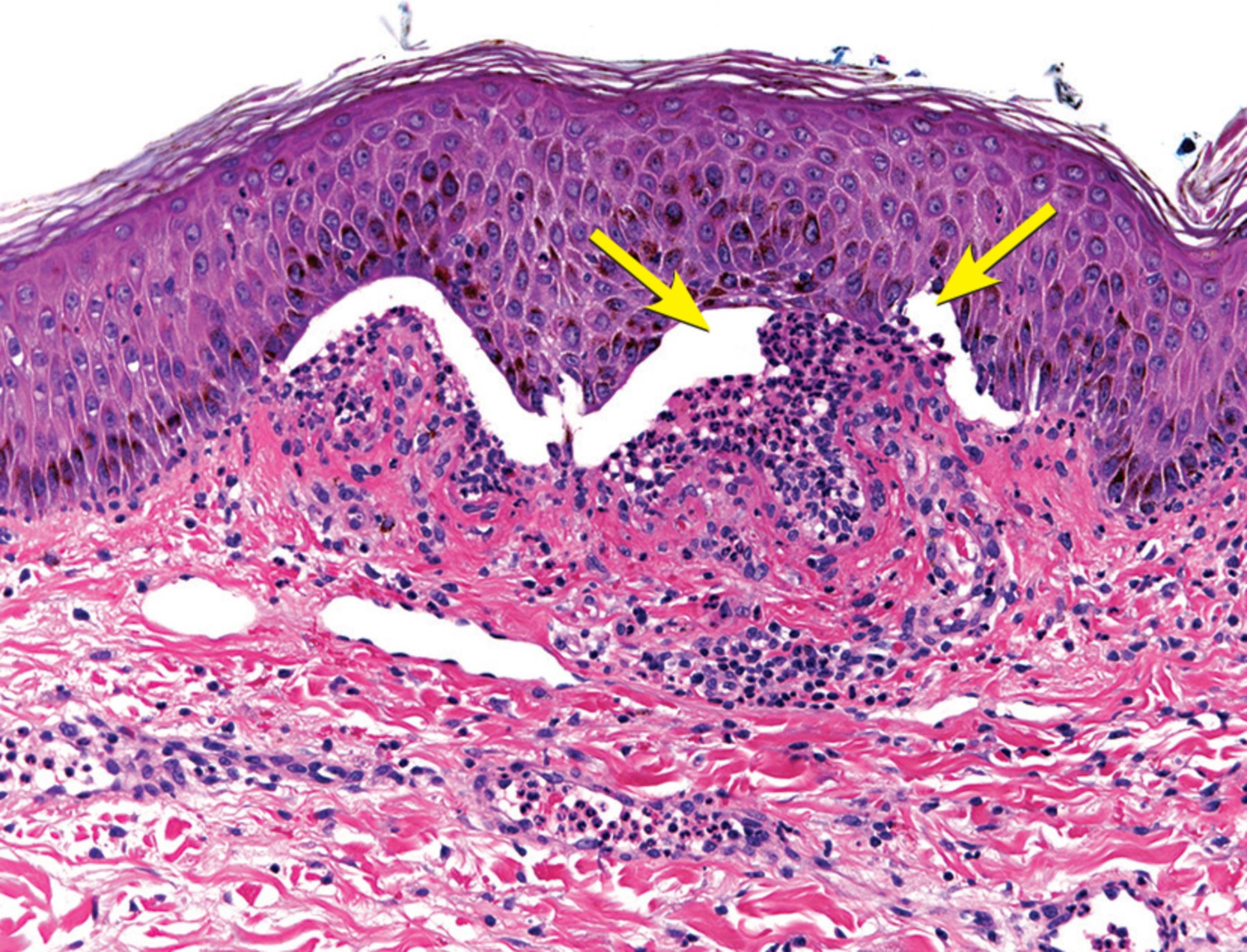
A skin biopsy of the left hand showed subepidermal bullae with numerous neutrophils in the superficial dermis (Figure 3). A second skin biopsy, on direct immunofluorescence testing, showed linear immunoglobulin G (IgG) and C3 at the dermoepidermal junction (Figure 4).

Biopsy from the left hand was significant for subepidermal bullae (arrows) with neutrophils.
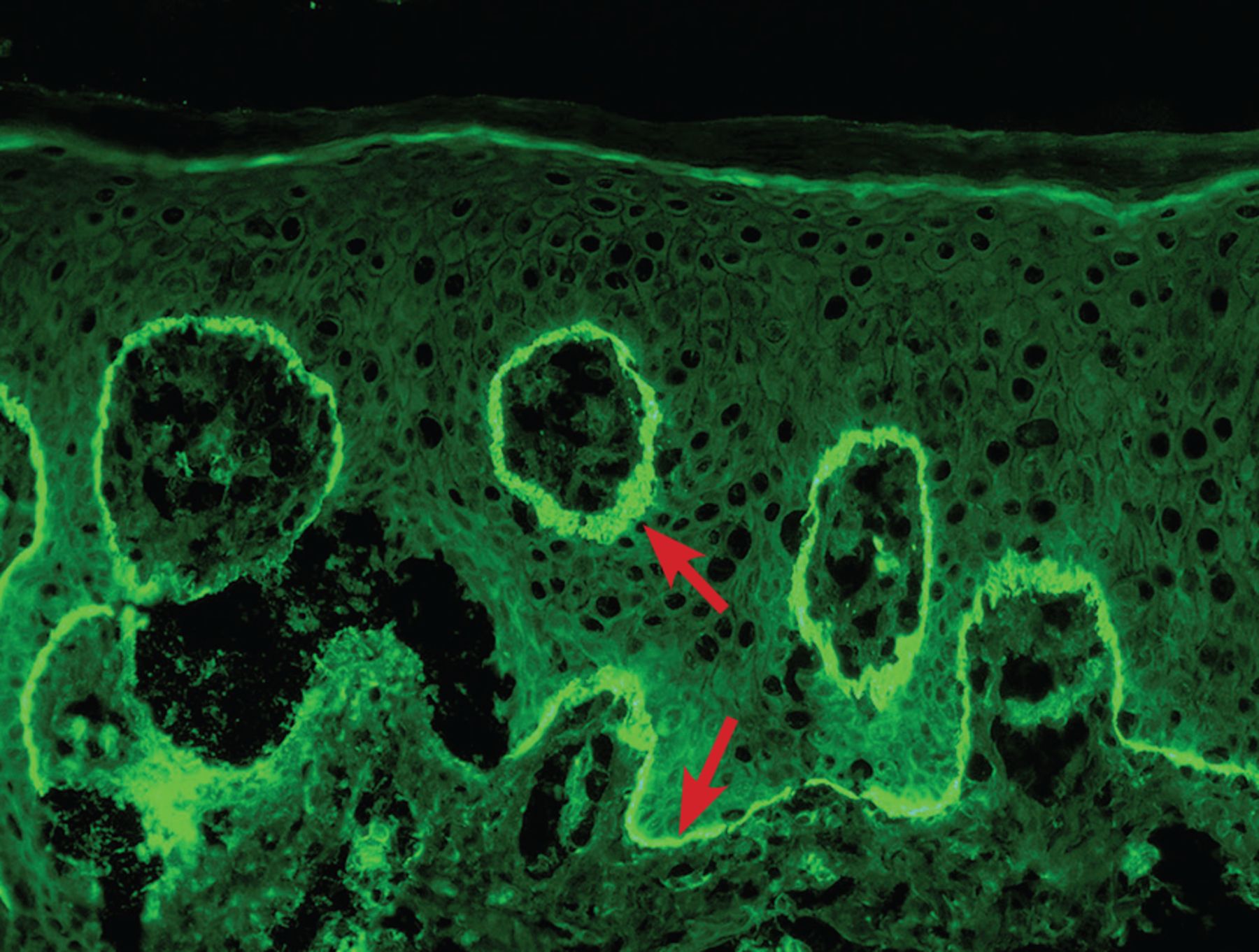
Immunofluorescence showed linear immunoglobulin G and C3 at the dermoepidermal junction (arrows).
DIFFERENTIAL DIAGNOSIS
1. What is the most likely cause of this patient’s symptoms?
Atypical coxsackievirus infection
Recurrent herpes virus
Bullous pemphigoid
Epidermolysis bullosa acquisita
The skin biopsy results showing linear IgG and C3 on direct immunofluorescence confirmed a diagnosis of bullous pemphigoid.
Bullous pemphigoid is the most common autoimmune bullous dermatosis, affecting individuals with a median age of 70.1 The pathogenesis involves formation of IgG autoantibodies against BP180 and BP230, which are components of hemidesmosomes that maintain dermoepidermal adhesion in stratified epithelia.2
Clinical features include tense bullae, often accompanied by erythematous or urticarial plaques on the abdomen, flexor surfaces of the extremities, axillae, or inguinal folds that may persist for days before developing erosions and crusts. Blisters may be preceded by an intensely pruritic prodrome,1 although this does not develop in at least 20% of cases.3 Mucosal involvement, as seen in this patient, occurs in 10% to 20% of patients.1
The technical standard for diagnosis is direct immunofluorescence revealing linear IgG or C3 deposits, or both, at the dermoepidermal junction on skin biopsy, though indirect immunofluorescence testing and enzyme-linked immunosorbent assay detection of BP180 or BP230 antibodies may help with the diagnosis.4
The most likely causes of our patient’s bullous pemphigoid are discussed in the following sections.
DRUG-INDUCED BULLOUS PEMPHIGOID
More than 50 medications have been associated with the development of bullous pemphigoid, including etanercept, sulfasalazine, furosemide, and penicillin.5–7 Drug-induced bullous pemphigoid may present acutely and resolve quickly after the offending agent is removed, or it may follow a chronic course that resembles idiopathic bullous pemphigoid. In either case, symptoms may develop up to 3 months after the medication was started.8
Although losartan has been linked to bullous pemphigoid in a single case,9 loop diuretics such as furosemide are among the most commonly reported culprits.5 There seems to be no association with the dose of furosemide and development of bullous pemphigoid, with doses in case reports ranging from 40 to 120 mg daily.10
Because there are no known antibodies specific to drug-induced bullous pemphigoid,11,12 it is possible that our patient coincidentally developed idiopathic bullous pemphigoid in the setting of furosemide therapy. However, her lesions improved after we stopped her furosemide while continuing losartan (see Management), supporting furosemide as the causative agent.
A 2020 systematic review found that in addition to loop diuretics, other agents with the greatest evidence supporting their role in drug-induced bullous pemphigoid are dipeptidyl peptidase 4-inhibitors (gliptins), inhibitors of programmed cell death protein 1 and programmed cell death ligand 1, and penicillin derivatives.13
Histopathology
Light microscopy of bullous pemphigoid lesions typically reveals subepidermal blisters with an eosinophil-rich superficial dermal infiltrate.1 Neutrophilic infiltrates, as seen in this patient’s biopsy, are rarely reported in bullous pemphigoid, and they are more typically associated with other bullous diseases such as linear IgA bullous dermatosis and dermatitis herpetiformis.14 Nonetheless, in this patient, the absence of linear IgA deposits at the dermoepidermal junction on immunofluorescence ruled out linear IgA bullous dermatosis.15
Although BP180 and BP230 antibodies were not detected in her serum, about 8% of bullous pemphigoid cases do not have significant levels of circulating BP180 autoantibodies,16 and 8% of people without bullous pemphigoid test positive for 1 or both autoantibodies.17
COXSACKIEVIRUS INFECTION
Coxsackievirus A16, a nonpolio enterovirus, causes hand, foot, and mouth disease (HFMD). This disease is most common in children under 5 years of age, but it may affect older children and adults.
Cases classically manifest as an oral enanthem or a nonpainful, nonpruritic exanthem of the hands, feet, buttocks, thighs, and arms. The enanthem is characterized by erythematous macules that progress to vesicles on erythematous bases before ultimately ulcerating. The exanthem may be macular, maculopapular, or vesicular, and most commonly arises on the dorsum of the hands and feet, occasionally affecting the palms and soles.
The diagnosis is clinical in children but may be confirmed by biopsy in adults, for whom the differential diagnosis is more extensive owing to consideration of autoimmune bullous diseases. Biopsy of HFMD lesions typically reveals loose strands of fibrin, lymphocytes, monocytes, and neutrophils with acantholysis of the epidermis.18
In the past decade, outbreaks of so-called atypical HFMD have been documented in adults with coxsackievirus A6. In contrast to typical HFMD, skin lesions in atypical HFMD are painful, are distributed more widely across the body, and may include bullae and eschar formation. Biopsy of atypical HFMD skin lesions reveals intense edema, necrotic keratinocytes, and neutrophilic exocytosis with T-cell infiltrate.19 In our patient’s case, biopsy results were not consistent with either typical or atypical coxsackievirus.
HERPES VIRUS INFECTION
Given this patient’s oral lesions and history of herpes labialis, erythema multiforme arising from HSV infection was included in the differential diagnosis. Herpes labialis typically presents as painful ulcerations at the vermilion border or the buccal mucosa that last up to 8 days and may be preceded by a painful or pruritic prodrome. It may be complicated by erythema multiforme, a self-limited cutaneous autoimmune disease characterized by targetoid lesions with 2 or 3 different concentric zones, with or without bullae formation in the center zone.
Erythema multiforme most commonly arises from HSV infection, but it may also be caused by drug reaction or other infectious pathogens including Mycoplasma pneumoniae, hepatitis C virus, Epstein-Barr virus, or coxsackievirus. Histologic features of erythema multiforme vary depending on the cause of the disease, site of biopsy within the skin lesion, and time point of biopsy in the disease course, but they generally include spongiosis, keratinocyte necrosis, and inflammatory infiltrate at the dermoepidermal junction.20 In patients with HSV-associated erythema multiforme, HSV DNA is detected in 43% of lesional skin biopsies.21 It was not detected in our patient’s skin lesions.
EPIDERMOLYSIS BULLOSA ACQUISITA
Epidermolysis bullosa acquisita (EBA) is a rare autoimmune blistering disorder characterized by the production of autoantibodies against type VII collagen, which anchors fibrils at the dermoepidermal junction and provides stability to structures in the extracellular matrixes. The clinical presentation varies but classically involves skin fragility and the formation of trauma-induced, noninflammatory, tense bullae in an acral distribution, with or without mucosal involvement.22
A subtype of EBA known as bullous pemphigoid-like EBA, which presents with tense bullae surrounded by inflamed or urticarial skin, was considered in this patient’s differential diagnosis given the skin lesions on her hands.
Up to 50% of patients with EBA have a bullous pemphigoid-like presentation, and in a review of sera from 85 patients diagnosed with bullous pemphigoid, 10% of bullous pemphigoid patients had circulating EBA antibodies.23 In our patient, the negative results for collagen type VII IgG antibodies excluded a diagnosis of EBA.
MANAGEMENT
2. What is the best next step in treating this patient’s bullous lesions?
Topical tacrolimus
Oral corticosteroids
Dapsone
Tetracycline plus nicotinamide
Discontinue furosemide
Furosemide was discontinued, and the patient was prescribed triamcinolone 0.1% cream to use on an outpatient basis. One month after discharge, she had only 2 urticarial plaques, and she was completely clear 2 months later.
Treatment with topical corticosteroids applied to affected areas and avoiding the face is often sufficient in mild, localized bullous pemphigoid regardless of the cause. Tapering the steroid dose may begin after 15 days of disease control, defined as the time point at which new bullous lesions and pruritus cease to form and existing lesions begin to heal, with a total treatment duration of 4 to 12 months. Supportive skin care such as baths containing antiseptics or wheat starch, or both, and application of nonadherent dressings to erosive lesions may improve the patient’s comfort, reduce bacterial infection, and promote wound healing.24 No large-scale clinical trial has evaluated the safety and efficacy of topical tacrolimus in bullous pemphigoid.
Severe or recurrent disease may warrant use of oral steroids, tetracyclines plus nicotinamide, dapsone, or immunosuppressive agents, and should be supervised by a dermatologist.24 In patients with drug-induced bullous pemphigoid, such as our patient, the suspected causative drug should be discontinued immediately.5
As a substitute for furosemide to treat her hypertension, the patient’s losartan dose was increased to 100 mg daily, and she was also prescribed amlodipine 10 mg daily and hydralazine 50 mg every 8 hours, with plans for close follow-up with her primary care physician. At discharge, her blood pressure had improved but remained elevated at 150/75 mm Hg. There is not enough literature available to guide the choice of an alternative to loop diuretics in patients with a history of furosemide-induced bullous pemphigoid.
FOLLOW-UP
3. Patients with bullous pemphigoid are more likely than patients without the disease to develop which of the following morbidities?
Kidney disease
Neurologic disorders
Malignancy
Heart disease
Our patient was found to have Alzheimer dementia 4 months after developing bullous pemphigoid, consistent with the documented association between bullous pemphigoid and neurologic disorders including dementia, stroke, epilepsy, Parkinson disease, and multiple sclerosis.25–27 While the mechanism of this association is unclear, immunologic cross-reactivity of antibodies targeting the BP230 isoforms in both the epidermis and brain may play a role.28,29
Retrospective studies have detected a higher rate of bullous pemphigoid in patients with laryngeal and renal cancers, as well as hematologic malignancies.30,31 However, a recent systematic review and meta-analysis that included these studies did not identify an association between bullous pemphigoid and malignancy.32
Although patients with bullous pemphigoid have more frequent hospitalizations and comorbidities,33,34 there is no documented association between bullous pemphigoid and heart or kidney disease in particular.
TAKE-HOME POINTS
If bullae develop in a patient taking furosemide, consider drug-induced bullous pemphigoid.
Bullous pemphigoid is diagnosed by a medical history, physical examination, and skin biopsy for evaluation by light microscopy and immunofluorescence.
In treating drug-induced bullous pemphigoid, stop the causative drug immediately, and apply topical corticosteroids.
Bullous pemphigoid is associated with neurologic disorders, including dementia.
DISCLOSURES
The authors report no relevant financial relationships which, in the context of their contributions, could be perceived as a potential conflict of interest.
Acknowledgments
The authors acknowledge and thank the patient discussed in this report.
- Copyright © 2021 The Cleveland Clinic Foundation. All Rights Reserved.




{kind=link}
{kind=link}
{kind=link}
{kind=link}